Visão geral da aprovação prévia à comercialização de dispositivos médicos USFDA
O processo de aprovação USFDA (PMA) USFDA é uma das vias de registo de dispositivos fornecidas pelaFDA US , projetada principalmente para dispositivos médicos FDA III FDA . O processo de aprovação FDA para dispositivos da Classe III envolve avaliações científicas e regulatórias meticulosas para avaliar a segurança e a eficácia do dispositivo médico, garantindo que os mais altos padrões sejam atendidos antes da autorização de comercialização.
Marque uma reunião com os nossos especialistas em aprovação antes da comercialização
Quem deve apresentar um submissão de aprovação pré-comercialização (PMA) de dispositivos USFDA ?
Os fabricantes de dispositivos devem apresentar uma submissão PMA submissão o dispositivo:
- É um romance.
- Pertence a uma classe de alto risco.
- Não pode ser encontrado na base de dados de classificação de produtos.
- Não é substancialmente equivalente (NSE) aos dispositivos das classes I, II ou III.
Obtenha aconselhamento especializado sobre submissão sua submissão de aprovação pré-comercialização
Qual é a diferença entre os pedidos 510(k), PMA e De-Novo?
Aprovação antes da comercialização
- Dispositivo pertencente à classe III que suporta a vida humana ou que apresenta um risco potencial e não razoável de doença ou lesão.
- O processo de aprovação de PMA FDA requer ensaios clínicos.
- Exige uma inspeção no local antes de emitir a aprovação da PMA.
- 180 dias de calendário
Classificação De-Novo
- Novos dispositivos das classes I e II que não tenham um dispositivo de referência válido.
- Requer dados de estudos clínicos.
- Não há auditoria no local antes da aprovação da De-Novo.
- 150 dias de calendário.
Registo 510(k)
- Dispositivos da classe III FDA que têm uma equivalência substancial com o dispositivo original.
- Não requer ensaios em seres humanos.
- Não há auditoria no local antes da autorização 510(k).
- 90 dias de calendário.
Quais são os diferentes submissão para aprovação FDA ?
Os fabricantes podem optar por qualquer um dos quatro (04) submissão PMA a seguir, que melhor se adequem ao seu dispositivo:
- PMA tradicional
- PMA modular
- Protocolo de desenvolvimento de produtos
- Isenção de dispositivos humanitários
Quais são os requisitos de dados para a aprovação prévia à comercialização de dispositivos médicos?
De acordo com a norma 21 CFR parte 814, os requerentes devem apresentar àFDA US um submissão CDRH devidamente preenchido, os compromissos exigidos e um dossiê técnico PMA bem elaborado. O dossiê técnico deve incluir os dados não clínicos e clínicos.
Dados não clínicos - Consistem em dados sobre microbiologia, toxicologia, imunologia, biocompatibilidade, stress, desgaste, prazo de validade e outros testes laboratoriais ou em animais.
Dados clínicos - Consistem em dados sobre protocolos de estudo, dados de segurança e eficácia, reacções adversas e complicações, falhas de dispositivos e substituições, informações sobre doentes, queixas de doentes, tabulações de dados de todos os sujeitos individuais, resultados de análises estatísticas e quaisquer outras informações das investigações clínicas.
O que é o submissão da PMA?
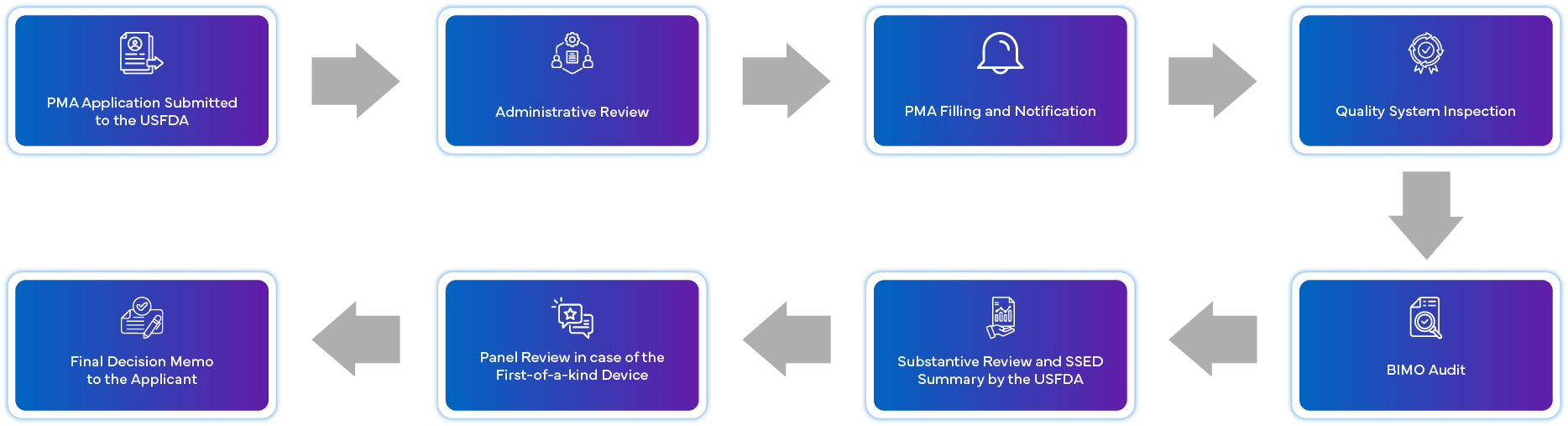
Quais são os requisitos de conformidade pós-aprovação para a PMA?
Os dispositivos aprovados ao abrigo da via da PMA devem cumprir os requisitos pós-comercialização estabelecidos pela USFDA. O dispositivo deve cumprir o seguinte:
- Requisitos pós-aprovação impostos na ordem de aprovação PMA FDA .
- Gestão de alterações pós-aprovaçãoatravés da apresentação atempada de suplementos de PMA relevantes
- Apresentação de relatórios pós-aprovação (anuais)
- Regulamentos 21 CFR 803 para Relatórios de Dispositivos Médicos (MDR)
- Estudos de vigilância pós-comercialização, conformeexigido pela USFDA nas ordens de aprovação de PMA.
Quais são as USFDA para analisar a submissão da PMA?
As taxas de utilização do MDUFA para PMA originais e suplementos são as seguintes
| submissão | suppara o ano fiscal de 2023 (sup de outubro de 2022 a sup de setembro de sup) | |
|---|---|---|
| Taxa normal | Taxa para pequenas empresas | |
| PMA, PDP, PMR, BLA | $441,547 |
|
| Suplemento à Panel-Track | $353,238 | $88,309 |
| Suplemento de 180 dias | $66,232 | $16,558 |
| Taxa anual para relatórios periódicos sobre um dispositivo da classe III (PMAs, PDPs e PMRs) | $15,454 | $3,864 |
| Aviso prévio de 30 dias | $7,065 | $3,532 |
| Suplemento em tempo real | $30,908 | $7,727 |
Com experiência no tratamento de submissões à PMA, a Freyr pode ajudar a identificar e compilar as informações, bem como auxiliar na preparação e revisão da submissão.
Experiência e vantagens USFDA aprovação prévia à comercialização de dispositivos médicos USFDA
- Diligência devida regulamentar
- Conformidade da inspeção do sistema de qualidade
- Conformidade da auditoria BIMO
- Compilação de ficheiros técnicos PMA
- Publicação e criação de eCopy
- Validação e apresentação da cópia eletrónica
- Aborda a resposta e as deficiências do RTA
- Serviços de ligação até à aprovação prévia ao mercado FDA
- Consulta de deficiências
- Listagem de dispositivos e registo de estabelecimentos
- Gestão de suplementos de PMA e avisos de 30 dias
- Apresentação de relatórios periódicos anuais
- Auditorias simuladas e 21 CFR 820 Formação

- Experiência no tratamento de muitas submissões de PMA FDA para categorias diversificadas de dispositivos.
- Equipa especializada para submissão de aprovação FDA , submissão com os requisitos regulamentares
- Apoio adicional para tratar de questões relacionadas com a PMA.
- Apresentação atempada dos resultados
- Em conformidade com asFDA alteraçõesFDA US
