2 min ler
O principal objetivoFDAUS é examinar constantemente e colmatar a lacuna entre os processos regulamentares para a importação e venda ininterruptas de dispositivos médicos novos e de alta qualidade no US .FDA 1998, aFDA US lançou um programa chamado «O novo paradigma 510(k): abordagens alternativas para demonstrar equivalência substancial nas notificações pré-comercialização». O objetivo é estabelecer um caminho eficiente para a submissão FDA (k) que contenha certas alterações na submissão 510(k) já aprovada. Esta nova notificação 510(k) oferece (03) três tipos de submissões, nomeadamente, 510(k) especial, 510(k) abreviada e 510(k) tradicional. Em 2019,FDA US emitiu um documento de orientação 510(k) especial que descreve um caminho opcional para fabricantes que fazem determinadas modificações bem definidas aos seus dispositivos comercializados legalmente.
Porquê um 510(k) especial?
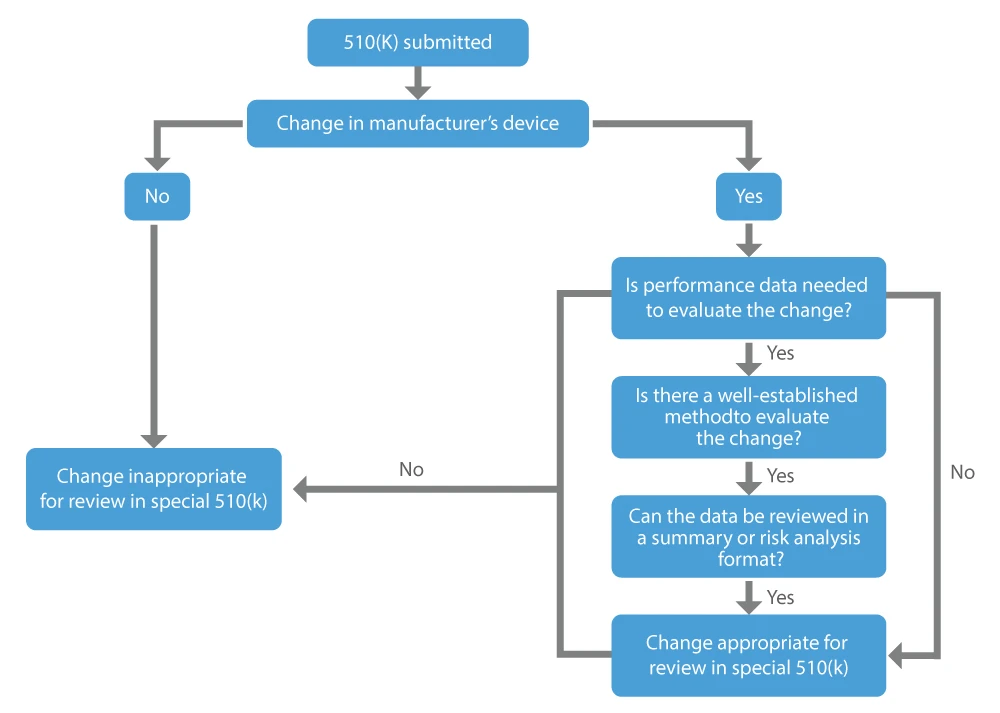
Quando um fabricante pretende obter aprovação para as modificações que efectuou no dispositivo já comercializado, ou seja, o dispositivo existente, pode solicitar uma autorização especial 510(k). Os principais factores a considerar ao determinar se uma alteração a um dispositivo existente pode ser adequada para um 510(k) especial são os seguintes:
- A alteração é efectuada no próprio dispositivo de referência legalmente comercializado pelo transmitente.
- Não são necessários dados de desempenho, ou estão disponíveis métodos bem estabelecidos, se forem considerados necessários para avaliar a alteração.
- Todos os dados de desempenho para apoiar uma determinação de equivalência substancial podem ser revistos num formato de resumo ou de análise de risco.
Documentos exigidos para a autorização especial 510(k)
- Carta de apresentação
- O nome do dispositivo legalmente comercializado (existente) do fabricante e o número 510(k)
- Uma descrição pormenorizada da(s) alteração(ões) efectuada(s) no dispositivo que levou à apresentação de um novo 510(k)
- Uma comparação do dispositivo modificado com o dispositivo limpo num formato tabular
- Outras alterações à rotulagem ou à conceção
- Um resumo conciso das actividades de controlo da conceção
- Com base na análise de risco, identificação das actividades de verificação e/ou validação necessárias para cumprir o disposto no 21 CFR 820.30
- Formulário de indicações de utilização
- Uma declaração de que o transmitente cumpriu e não viola atualmente os requisitos do procedimento de controlo da conceção, tal como especificado no 21 CFR 820.30 ( 21 CFR 820, e que os registos estão disponíveis para análise mediante pedido
Cronograma especial de revisão 510(k) pelaFDA US
De acordo com as diretrizes da FDA"Refuse to Accept Policy for 510(k)s", o prazo de revisão das apresentações especiais 510(k) é de trinta (30) dias após a sua receção.
Quando solicitar um pedido especial 510(k)?
FDA US FDA esforços consistentes para fornecer dispositivos médicos seguros e eficazes para promover a saúde humana. O programa especial 510(k) é eficiente e consistente com o procedimento de revisão menos oneroso, que ajuda os fabricantes estrangeiros a vender os seus dispositivos nos EUA e permite que os pacientes tenham acesso oportuno a novos dispositivos médicos.
Para qualquer esclarecimento adicional sobre o processo especial 510(k) da FDA, reach a Freyr - um especialista comprovado em Regulamentação. Mantenha-se informado. Mantenha-se em conformidade.