Übersicht über Medizinprodukte-Kombinationsprodukte
In der dynamischen Welt des Gesundheitswesens und der Innovation sind Medizinprodukt-Kombinationsprodukte zu einer starken Brücke geworden, die Pharmazeutika, Medizinprodukte und Biologika miteinander verbindet. Der Markt für Kombinationsprodukte wächst rasant, mit einer erwarteten jährlichen Wachstumsrate (CAGR) von 8,9 % von 2023 bis 2030. Der Sektor der Arzneimittel-Medizinprodukt-Kombinationsprodukte steht vor einem nachhaltigen Wachstum, das durch technologische Fortschritte, eine verbesserte Gesundheitsinfrastruktur, reibungslosere Regulierungswege, strategische Kooperationen und das Engagement für eine patientenzentrierte Versorgung gefördert wird.
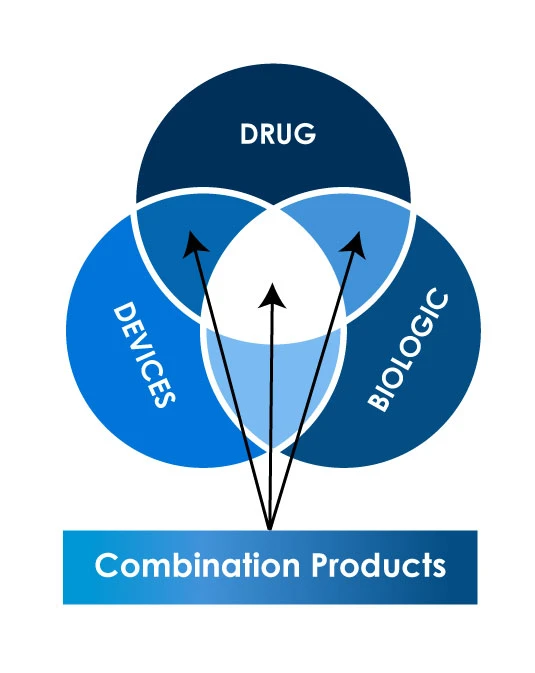
Verschiedene Arten von Kombinationsprodukten
Globales Regulierungsszenario für die Registrierung von Kombinationsprodukten
Die Auslegung dessen, was ein Kombinationsprodukt darstellt, kann von Land zu Land variieren, was die Komplexität der Registrierung solcher Produkte in verschiedenen Ländern erhöht. Darüber hinaus können die regulatorischen Anforderungen und Verfahren für Kombinationsprodukte Unterschiede in Dokumentation, Kommunikation und Validierung aufweisen. Die regulatorische Landschaft für die Registrierung von Kombinationsprodukten kann weltweit erheblich variieren. Hier sind die wichtigsten Regulierungsbehörden, die diese Produkte weltweit überwachen.
| Land | Behörde | Federführende Genehmigungszentren |
|---|---|---|
| USA | Büro für Kombinationsprodukte (OCP) | Center of Drug Evaluation and Research (CDER) |
| Zentrum für die Bewertung und Forschung biologischer Produkte (CBER) | ||
| Center of Devices and Radiological Health (CDRH) | ||
| EU | Benannte Stellen (NB) | Nationale zuständige Behörde (Arzneimittel) |
| Benannte Stellen (NB) (Medizinprodukte) | ||
| Japan | Abteilung für Bewertung und Zulassung oder das Amt für Medizinprodukte/zelluläre und gewebebasierte Produkte des Amtes für Pharmazeutische und Lebensmittelsicherheit | Direktor der Abteilung für Bewertung und Zulassung (DMDL), Amt für Pharmazeutische und Lebensmittelsicherheit, Amt für Pharmazeutische und Medizinische Sicherheit, Ministerium für Gesundheit und Soziales |
| China | Zentrum für die Verwaltung der Medizinprodukte-Standardisierung (CMDSA) | Center of Medical Device Evaluation (CMDE) |
| Zentrum für Arzneimittelbewertung (CDE) | ||
| Malaysia | Nationale Regulierungsbehörde für Pharmazeutika | Nationale Pharmazeutische Regulierungsbehörde (NPRA) |
| Medizinprodukteagentur |
Die Registrierung von Kombinationsprodukten auf internationalen Märkten erfordert einen maßgeschneiderten Ansatz, der eine enge Zusammenarbeit mit den zuständigen Gesundheitsbehörden zur Genehmigung beinhaltet. Der typische Prozess für die Registrierung von Kombinationsprodukten umfasst die folgenden Schritte:
- Bewertung, ob ein bestimmtes Produkt die Kriterien für die Klassifizierung als Kombinationsprodukt erfüllt.
- Klassifizierung von Produkten basierend auf den damit verbundenen Risiken.
- Identifizierung der relevanten Standards und Datenanforderungen, die von der jeweiligen Gesundheitsbehörde festgelegt wurden.
- Erstellung der notwendigen Daten gemäß den Vorgaben der Behörde.
- Erstellung einer technischen Dokumentation gemäß den spezifischen Anforderungen jedes Landes.
- Einreichung des Antrags und Beantwortung von Anfragen oder Bedenken bis zur Erteilung der Genehmigung.
- Management des Produktlebenszyklus nach der Zulassung.
Unsere Kompetenzen
- Erste Risikoanalyse
- Marktforschung – Produktspezifische Markteinblicke
- Personalaufstockung
- Erstellung regulatorischer Strategien.
- Potenzielle Märkte und Zulassungswege.
- Entwicklungsakte und Risikoanalyse.
- Qualitätsmanagementsystem (QMS) ISO 13485
- Medical Device Single Audit Program (MDSAP)
- QMS ISO 13485 Vorab-Bewertung
- Regulatorische Strategie
- Freyr IMPACT (Plattform für regulatorische Informationen)
- Verifizierung und Validierung des Designs
- Risikomanagement
- Entwurf der technischen Dokumentation
- Regulatorische Strategie
- Regulatorische Anforderungen
- Freyr rDMS-Tool (Daten-/Dokumentenmanagementsystem)
- Prozess- und klinische Validierung
- Endgültige Kennzeichnung und Artwork
- Landesvertretung
- Regulatorische Einreichung
- Die CE-Kennzeichnung (Conformité Européenne) der Europäischen Union (EU) und die UKCA-Kennzeichnung (UK Conformity Assessment)
- Zertifizierung für den globalen Marktzugang
- Audit-Unterstützung durch Benannte Stellen (NB) / Zugelassene Stellen
- Landesvertretung
- Regulatorische Zulassungen
- Überwachung nach dem Inverkehrbringen (PMS)
- PMCF (Post-Market Clinical Follow-up)
- Jährliche Pflege der technischen Dokumentation (CER/Risikomanagement)
- Erneuerung von Zulassungen
- Neue Markteinführungen
- Kommunikation mit der Zuständigen Behörde / Benannten Stelle / Zugelassenen Stelle
- Automatisierte Pharmakovigilanz (PV)-Lösungen
Warum Freyr?
Registrierung von Medizinprodukten
- Umfassende Regulierungsstrategie für Kombinationsprodukte.
- Regulatorische Unterstützung für Produktentwicklungsdokumente wie Design History Files (DHFs).
- QMS-Konformitätsstrategie.
- Regulatorische Konformität, Lückenanalyse und Behebung von Mängeln in technischen Dokumenten und Qualitätssystemen.
- Dienstleistungen für regulatorische Kennzeichnung und technische Redaktion.
- Dienstleistungen für regulatorische und Marktinformationen.
- Übersetzungsdienstleistungen für Dokumente und Kennzeichnungen.
- Kontakt und Betreuung bei Gesundheitsbehörden.
- Dienstleistungen für regulatorisches Artwork.
- Pharmakovigilanz- und PMS-Dienstleistungen.
- Publikationsdienstleistungen.
- Medizinische Redaktionsdienstleistungen.

- Erfolgreiche Einreichungen für verschiedene Klassen von IVD.
- Engagiertes und erfahrenes Personal zur Bereitstellung regulatorischer Unterstützung für Medizinprodukte und IVD.
- Pünktliche Einreichung von Leistungen.
- Zugang zu lokalen Partnern, um den Herausforderungen der Behörden und sprachspezifischen Anforderungen gerecht zu werden.
- Unterstützung durch einen lokalen oder gesetzlichen Vertreter mit einem kosteneffizienten Modell.
- Management regulatorischer Ressourcen / Dienstleistungen zur Personalergänzung.