US FDA Übersicht zur Medizinprodukte-Registrierung
Die Vereinigten Staaten von Amerika (USA) sind bekannt als stark regulierter Markt für Medizinprodukte mit klar definierten Registrierungswegen und -anforderungen. Die ursprünglichen Medizinproduktevorschriften der USA stammen aus dem Jahr 1976 und haben sich im Laufe der Zeit weiterentwickelt. Sie werden vom Centre for Devices and Radiological Health (CDRH) unter der Food and Drug Administration (FDA) reguliert. Freyr hat zahlreichen Geräteherstellern geholfen, den Registrierungsprozess für Medizinprodukte der US FDA einzuhalten.
![]()
Aufsichtsbehörde: Lebensmittel- und Arzneimittelbehörde (FDA)![]()
Verordnung: Titel 21 Code of Federal Regulations (21 CFR) Teile 800 – 1299![]()
Regulatorischer Weg: Pre-Market Notification oder Zulassung vor dem Inverkehrbringen oder De-Novo-Klassifizierung![]()
Bevollmächtigter Vertreter: US Agent![]()
QMS-Anforderung: Qualitätssystem-Verordnung (QSR) (21 CFR Teil 820)![]()
Bewertung technischer Daten: Centre for Devices and Radiological Health![]()
Gültigkeit der Lizenz: Unbegrenzt![]()
Kennzeichnungspflichten: 21 CFR Teil 801![]()
Einreichungsformat: Papier & CD/DVD![]()
Sprache: Englisch
Medizinprodukteklassifizierung US
Die FDA klassifiziert Medizinprodukte in drei risikobasierte Kategorien: Klasse I, Klasse II und Klasse III. Geräte der Klasse I gelten als risikoarm, während Geräte der Klasse III mit hohem Risiko verbunden sind. Die Registrierungsanforderungen und der Zulassungsweg variieren je nach Geräteklasse.
| Geräteklasse | Risiko | Registrierungsweg zur Zulassung |
|---|---|---|
| I | Geringes Risiko | 510(k)-befreit |
| II | Mittleres Risiko (Mit Vergleichsprodukt) | Vormarktbenachrichtigung/510(k) |
Mittleres Risiko (Ohne Vergleichsprodukt) | De-Novo-Antrag | |
| III | Hohes Risiko | Vormarktzulassung (PMA) |
US FDA-Beauftragter
Unternehmen ohne lokale Niederlassungen in den US müssen einen US FDA Agenten ernennen, um den Hersteller zu vertreten. Der US FDA Agent muss entweder in den US ansässig sein oder einen Geschäftssitz in den US unterhalten. Die vom Agenten zu erfüllenden Aufgaben sind von der US FDA als Teil der CFR-Vorschriften vorab festgelegt.
Navigieren Sie durch die Häufig gestellten Fragen (FAQs) zum US-Agenten.
Interaktive Besprechungen mit der US FDA
Die US FDA unterstützt Hersteller durch verschiedene Arten von Q-Submission-Meetings, um unterschiedliche Ziele zu erreichen. Solche Treffen mit der Behörde vor Beginn oder während der Entwicklung eines Medizinprodukts, noch vor der Einreichung von US FDA Anträgen zur Registrierung von Medizinprodukten, helfen Herstellern, Zeitpläne und Kosten für die Kommerzialisierung des Produkts zu optimieren.
Registrierung von Medizinprodukten US
Die Geräte können vom CDRH, FDA über verschiedene Registrierungswege zugelassen werden. Diese sind:
Medizinprodukte der Klasse I: Geräte der Klasse I sind in der Regel von der GMP- und 510(k)-Einreichung befreit und benötigen keine vorherige Genehmigung der US FDA, um sie in den US zu vermarkten. Andere Anforderungen wie die Registrierung der Betriebsstätte, die Produktlistung, UDI, PMS usw. müssen vom Hersteller eingehalten werden.
Medizinprodukte der Klasse II: Mittelrisikogeräte mit 510(k)-zugelassenen Vergleichsgeräten können sich für eine 510(k) Pre-market Notification (PMN) entscheiden, auch bekannt als 510(k)-Registrierung. Das betreffende Gerät muss eine wesentliche Äquivalenz (SE) zu den identifizierten und beanspruchten Vergleichsgeräten nachweisen. Dieser Weg ist der am weitesten verbreitete Weg zur Registrierung von Geräten in den US. Hersteller von Mittelrisikogeräten ohne Vergleichsgeräte können eine Klassifizierung durch die US FDA über De-Novo-Anträge beantragen.
Medizinprodukte der Klasse III: Hersteller von Hochrisikogeräten der Klasse III müssen einen Pre-Market Approval (PMA)-Antrag bei der US FDA einreichen. Die Geräte müssen einer detaillierten klinischen Bewertung unterzogen werden, und der Hersteller muss detaillierte Sicherheits- und Wirksamkeitsdaten aus klinischen Studien vorlegen. Die US FDA führt im Rahmen der Bewertung eine QMS-Inspektion durch, bevor sie eine Pre-Market Approval für das Gerät erteilt.
Registrierungen von Medizinprodukten, die nicht unter das CDRH fallen
Basierend auf den Verwendungszwecken beziehen einige Grenzprodukte, die in anderen Ländern als Medizinprodukte gelten, wie chirurgische Atemschutzmasken, Desinfektionsmittel und Kombinationsprodukte, andere Behörden ein, darunter das Centre for Disease Control (CDC), das National Institute for Occupational Safety and Hazards (NIOSH), die Environmental Protection Agency (EPA), das Centre for Biological Evaluation and Research (CBER) und das Centre for Drug Evaluation and Research (CDER).
Anforderungen an die Einhaltung von Vorschriften nach der Zulassung für Medizinprodukte
Alle Hersteller von Medizinprodukten müssen die unten aufgeführten Anforderungen nach der Zulassung einhalten:
- Anforderung an Registrierung und Listung: Einrichtungen aller Produktklassen müssen in der FURLs-Datenbank registriert werden, und das Produkt muss gelistet werden, nachdem die Zulassung erteilt wurde und bevor es in den US vermarktet wird. Einige Produkte, wie z.B. Strahlungsgeräte, müssen weitere Anforderungen, wie z.B. eine Zugangsnummer, erfüllen, bevor sie in die US importiert werden können.
- Eindeutige Produktidentifikation: Alle Produktklassen müssen die Vorschriften zur eindeutigen Produktidentifikation (UDI) einhalten, um die Produkte in den US zu vermarkten.
- Registrierungsgebühren: Der Hersteller muss die jährlichen Registrierungsgebühren zahlen, um seine Registrierung aktiv zu halten und weiterhin Produkte in den US zu vermarkten. Die US FDA hat eine reduzierte Gebührenstruktur für kleinere Unternehmen mit einem aktiven Small Business Certificate.
- Qualitätsaudits: Für Produkte, die nicht von der GMP befreit sind, kann die US FDA die Fertigungsstätte jederzeit auf Einhaltung der Qualitätsmanagementsystem-Vorschriften (QSR) gemäß der 21 CFR 820 inspizieren.
Prozessablauf
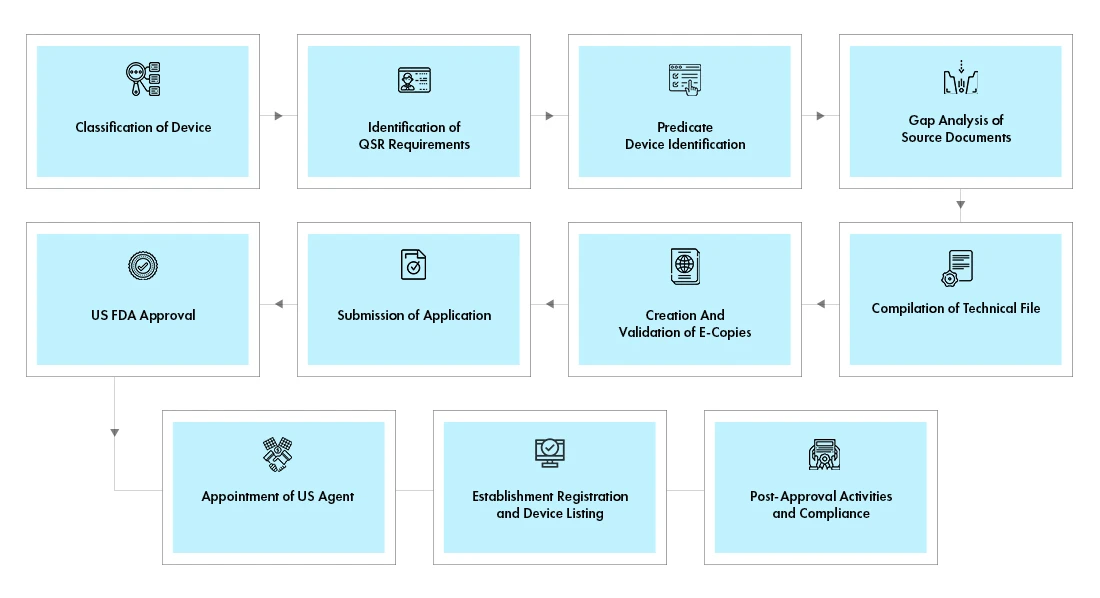
Lebenszyklusmanagement von Medizinprodukten nach der Zulassung
Freyr unterstützt ausländische Hersteller beim End-to-End Lebenszyklusmanagement von Medizinprodukten, einschließlich Aktivitäten nach der Zulassung, wie zum Beispiel:
- Änderungsmanagement nach der Zulassung – Änderungen an bestehenden Medizinprodukte-Zulassungen, wie z.B. die Hinzufügung neuer Varianten, Zubehörteile; die Hinzufügung neuer Anwendungsgebiete und Ähnliches
- Aufrechterhaltung von Zulassungen und Registrierungen durch fristgerechte Zahlung der MDUFA-Gebühren an die FDA
- Koordination zwischen der US FDA und dem Hersteller.
Freyr verfügt über ein exklusives Lieferzentrum in den US mit einem professionellen Team, das Herstellern regulatorische Unterstützung bei der Aufrechterhaltung der für die Zulassung erforderlichen Qualität und Sicherheit bietet. Die Experten von Freyr beobachten aufmerksam regulatorische Aktualisierungen und informieren die Kunden über die notwendigen Schritte zur Produktkonformität mit dem aktuellen Standard.
Zusammenfassung
| Risiko | Geräteklasse | QMS-Audit | Verfügbarkeit von Referenzprodukten | Regulatorischer Weg | US Agent | US FDA-Fristen |
|---|---|---|---|---|---|---|
| Geringes Risiko | I | Nein | N/A | Ausgenommen | Ja | 1 Monat |
| Mittleres Risiko | II | Ja (nach Zulassung) | Ja | PMN/510(k) | Ja | 9 - 12 Monate |
| Mittleres Risiko | II | Ja (nach Zulassung) | Nein | De-Novo-Klassifizierungsantrag | Ja | 18 - 30 Monate |
| Hohes Risiko | III | Ja (vor der Zulassung) | N/A | PMA | Ja | 18 - 30 Monate |
Dienstleistungen von Freyr zur Registrierung von Medizinprodukten
Freyr Expertise
- Regulatorische Due-Diligence
- Produktdokumentation
- 513(g) Unterstützung
- 510(k)-Registrierung
- De-Novo-Antrag auf Klassifizierung
- PMA-Registrierung
- 21 CFR 820-Konformität
- BIMO Audit Unterstützung
- MDSAP Konformität
- Kennzeichnungsunterstützung
- Unterstützung bei Veröffentlichung und Einreichung
- US Agent
- Q-Submission-Besprechungen
- RFD- und Pre-RFD-Besprechungen
- Zertifizierung als Kleinunternehmen
- Betriebsregistrierung und Gerätelistung
- Regulatorische Konformität für Medizinprodukte mit Strahlung
- Änderungsmanagement nach der Zulassung
- Post-Market Surveillance
- UDI-Konformität
- Regulatorische Beratung zur Behebung von Mängeln