


Übersicht zur Registrierung von Medizinprodukten in Indonesien
Indonesien hat 2014 eine allgemeine Gesundheitsversorgung für seine Bürger eingeführt. Dies hat das Wachstum des Marktes für Medizinprodukte stark beeinflusst und zu einem Anstieg der Importe von Medizinprodukten geführt. Die Medizinprodukte in Indonesien werden von der Nationalen Agentur für Arzneimittel- und Lebensmittelkontrolle (NADFC) reguliert, die dem indonesischen Gesundheitsministerium (MoH) untersteht. Die neueste geltende Verordnung für den Import von Medizinprodukten ist das Dekret Nr. 62, das im Jahr 2017 erlassen wurde. Ausländische Unternehmen müssen einen lokalen bevollmächtigten Vertreter in Indonesien für den Prozess der Registrierung von Medizinprodukten in Indonesien benennen.
![]()
Aufsichtsbehörde: Nationale Agentur für Arzneimittel- und Lebensmittelkontrolle (NADFC)![]()
Verordnung: Nr. 62 / 2017![]()
Bevollmächtigter Vertreter: Lokaler bevollmächtigter Vertreter in Indonesien![]()
QMS-Anforderung: ISO 13485:2016![]()
Bewertung technischer Daten: NADFC![]()
Gültigkeit der Lizenz: 5 Jahre![]()
Kennzeichnungspflichten: Nr. 62 / 2017![]()
Einreichungsformat: Online/Papier![]()
Sprache: Englisch & Indonesisch
Klassifizierung von Medizinprodukten in Indonesien
Die aktuelle Verordnung klassifiziert die Produkte je nach Risiko in die Klassen A, B, C und D.
| Risikokriterien | Geräteklasse |
|---|---|
| Geringes Risiko | A |
| Niedrig-moderates Risiko | B |
| Mittel – Hohes Risiko | C |
| Hohes Risiko | D |
Lokaler Bevollmächtigter in Indonesien
Die indonesischen Vorschriften verlangen von Herstellern, einen lokalen Bevollmächtigten mit einer Vertriebslizenz zu benennen. Ein Distributor kann ernannt werden, um den ausländischen Hersteller in Indonesien zu vertreten. Die Ernennung eines unabhängigen Dritten würde jedoch die Flexibilität bieten, Distributoren zu wechseln oder mehrere Distributoren für eine bessere Marktdurchdringung zu ernennen.
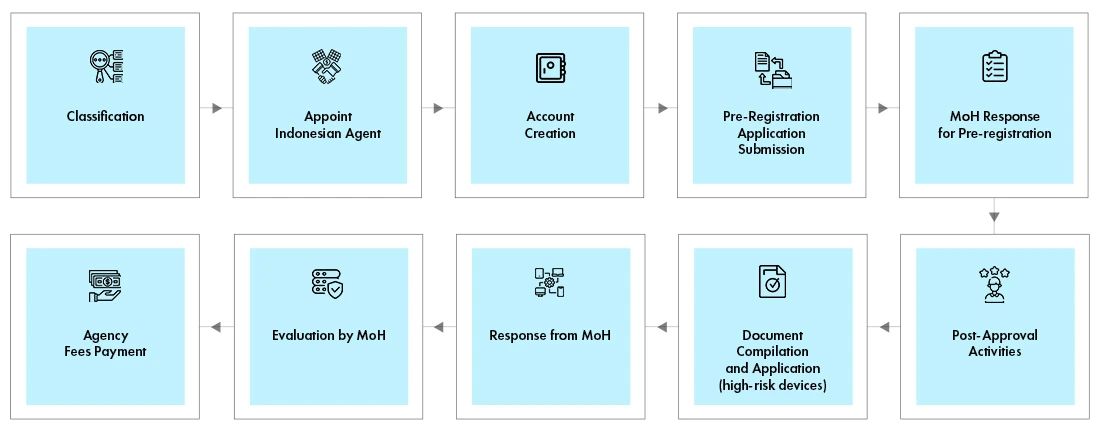
Registrierung von Medizinprodukten in Indonesien
Der lokale Bevollmächtigte muss ein Konto im Online-Portal erstellen. Der Registrierungsprozess ist für alle Produktklassen gleich. Die Dokumentationsanforderungen variieren jedoch je nach Produktklasse. Die Registrierung ist ein zweistufiger Prozess –
- Vorregistrierungsprozess
- Evaluierungsprozess
Das Gesundheitsministerium (MoH) überprüft die Klassifizierung des Produkts und legt die Kosten für die Evaluierung fest. Das Ergebnis der Vorregistrierung wird zusammen mit der Rechnung per E-Mail an den Antragsteller gesendet. Der lokale Bevollmächtigte nimmt im Namen des Herstellers die Zahlung vor und lädt den Zahlungsnachweis hoch. Das MoH prüft die Dokumente und teilt dem Antragsteller die Ergebnisse per E-Mail mit. Einige Produkte erfordern Tests im Land in einem akkreditierten Labor.
Übersicht über den behördlichen Genehmigungsprozess
Das Expertenteam von Freyr verfolgt die sich ändernden Trends und Vorschriften und unterstützt die Beteiligten bei der Aufrechterhaltung der regulatorischen Konformität über den gesamten Produktlebenszyklus hinweg. Wir bieten regulatorische Lösungen, um andere regulatorische Aspekte der Konformität innerhalb begrenzter Budgets zu gewährleisten.
Geräteklasse | Risikoklasse | Zeitpläne des Gesundheitsministeriums für Marktzulassung | Zeitpläne des Gesundheitsministeriums für Verlängerung / Änderung | ||
|---|---|---|---|---|---|
| Klassifizierungsprozess (Tage) | Bewertungsprozess (Tage) | Klassifizierungsprozess (Tage) | Bewertungsprozess (Tage) | ||
| Klasse A | Geringes Risiko | 7 | 45 | 7 | 45 |
| Klasse B | Niedrig-moderates Risiko | 7 | 90 | 7 | 45 |
| Klasse C | Mittel – Hohes Risiko | 7 | 100 | 7 | 45 |
| Klasse D | Hohes Risiko | 7 | 120 | 7 | 45 |
Freyr Expertise
- Regulatorische Due-Diligence
- Registrierung von Medizinprodukten
- Prüfung im Inland
- Händlerlizenzierung
- Legalisierung und Notarisierung
- Rechtlicher Vertreter
- Etikettierungsunterstützung
- Übersetzungsunterstützung
- Identifizierung und Qualifizierung von Vertriebspartnern
- Dienstleistungen für die Post-Market Surveillance
- Änderungsmanagement nach der Zulassung
- Dienstleistungen für die Lizenzverlängerung und -übertragung
- Einreichungs- und Vermittlungsdienstleistungen
