
Übersicht zur Medizinprodukte-Registrierung in Israel
Die israelische Medizinprodukteindustrie verzeichnet ein anhaltendes Wachstum und Innovation, was sie zu einem Zentrum für Spitzentechnologien im Gesundheitswesen macht. Die Registrierung von Medizinprodukten ist für Unternehmen, die in diesen dynamischen Markt eintreten, entscheidend. Dieser Überblick beleuchtet wichtige Aspekte des Registrierungsprozesses in Israel und bietet Einblicke in die regulatorischen Rahmenbedingungen und Anforderungen, um innovative Medizinprodukte im israelischen Gesundheitssektor zu etablieren.
Regulierungsbehörde: Die Abteilung für Medizinprodukte des israelischen Gesundheitsministeriums (AMAR).
Regulierung: Medizinprodukte-/Ausrüstungsgesetz von 2012
Regulatorischer Weg: Produktregistrierung
Lokaler autorisierter Vertreter in Israel: Israel Registration Holder (IRH)
QMS-Anforderung: ISO 13485
Bewertung technischer Daten: Die Abteilung für Medizinprodukte im Gesundheitsministerium
Gültigkeit der Lizenz: Fünf (05) Jahre
Einreichungsformat: Papier und Elektronisch
Übersetzung: Übersetzte Dokumente auf Hebräisch
Geräteklassifizierung
Das Gesetz über medizinische Geräte und die Vorschriften für die Registrierung medizinischer Geräte in Israel legen kein spezifisches Risikoklassifizierungssystem fest. Stattdessen richtet Israel seine Klassifizierung von Medizinprodukten an internationalen Standards aus, insbesondere an denen, die von den Ländern der Global Harmonization Task Force (GHTF) festgelegt wurden. Alternativ wird die Risikoklassifizierung eines Produkts aus einem anerkannten Land für die Registrierung in Israel übernommen. Dieser Klassifizierungsprozess berücksichtigt typischerweise den Verwendungszweck, das Risikoniveau und andere Faktoren, die die Sicherheit und Wirksamkeit von Medizinprodukten beeinflussen können.
Medizinprodukteklassen
| Klasse | Risiko |
|---|---|
| Klasse I | Niedrig |
| Klasse II | Niedrig bis Mittel |
| Klasse III | Hoch |
Vorgeschlagene Änderungen an den Registrierungswegen
Vorgeschlagene Änderungen betreffen Produkte der Klasse I und Klasse II, während das Registrierungssystem für Produkte der Klasse III unverändert bleibt.
- Produkte der Klasse I können sofort durch Selbsterklärung registriert werden.
- Für Medizinprodukte der Klasse II sind zwar Erklärungen und technische Dokumente erforderlich, AMAR kann den Prozess jedoch für Produkte mit geringem bis mittlerem Risiko auf vierzehn (14) Tage beschleunigen. Dies gilt, wenn der Hersteller zwei (02) Genehmigungen aus anerkannten Ländern besitzt und sechs (06) Monate Marktdaten vorlegt. Alternativ wird für Medizinprodukte der Klasse II, die nur die US FDA 510(k)-Zulassung und sechs (06) Monate US-Marktdaten vorweisen können, die AMAR-Bearbeitungszeit auf sechzig (60) Tage beschleunigt.
Lokaler autorisierter Vertreter in Israel
Medizinprodukteunternehmen mit Sitz außerhalb Israels müssen einen Israeli Registration Holder (IRH) benennen, um die Registrierung ihrer Produkte für den Verkauf im Land zu erleichtern. Der IRH fungiert als lokaler Vertreter des Herstellers und ist dafür zuständig, mit dem Gesundheitsministerium in Kontakt zu treten, um die Einhaltung der lokalen Vorschriften sicherzustellen. Darüber hinaus ist ein IRH für den Aufbau und die Aufrechterhaltung einer Geschäftspräsenz in Israel sowie für den Erwerb und die Pflege einer gültigen Geschäftslizenz verantwortlich.
Registrierung von Medizinprodukten
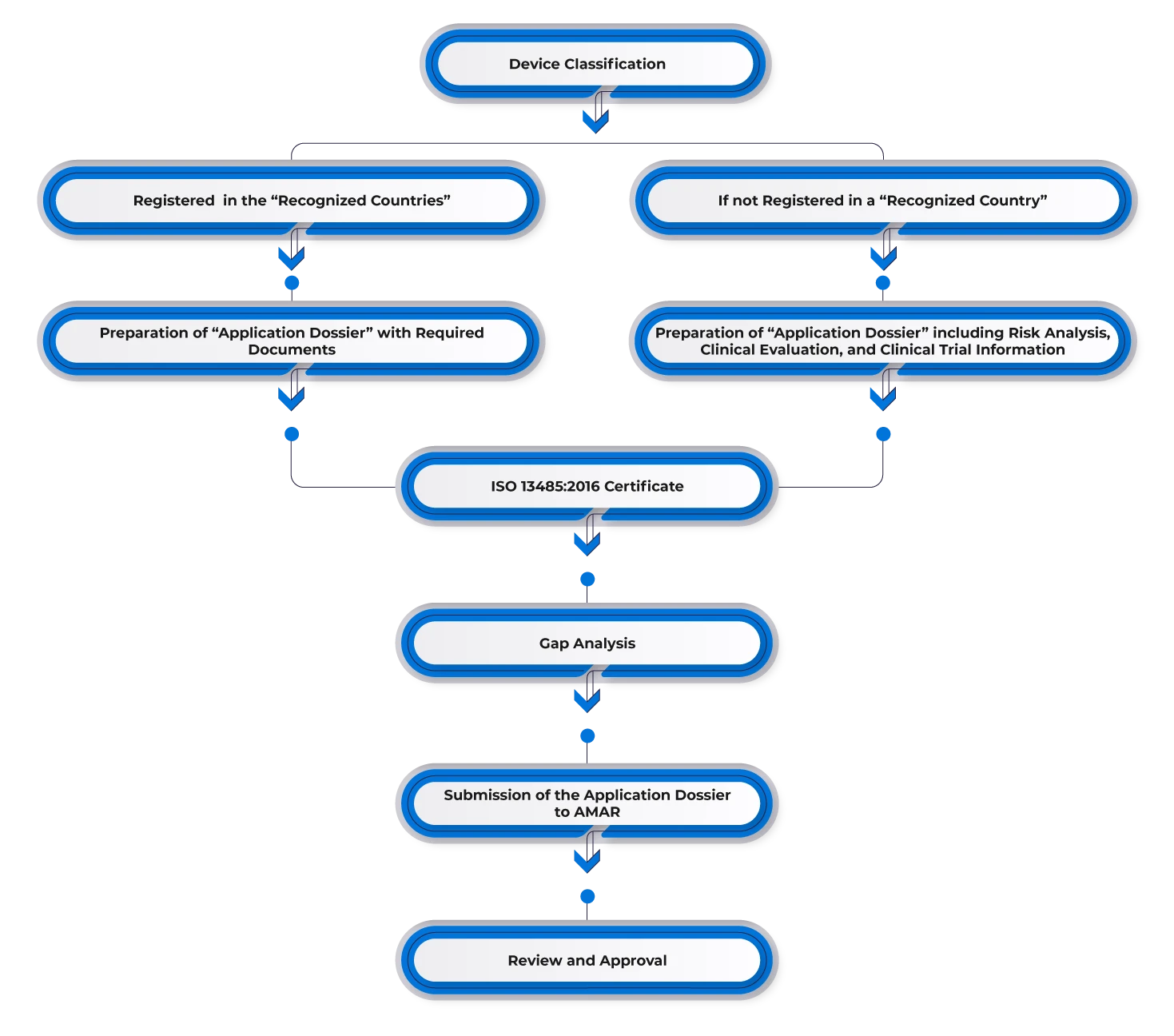
Um ein Medizinprodukt in Israel zu registrieren, müssen Hersteller eine vorherige Genehmigung in einem der Referenzmärkte wie den US, Europa, Australien, Kanada oder anderen wichtigen Märkten einholen. Hersteller mit bestehenden Genehmigungen in einem der Referenzländer können diese Genehmigung für den israelischen Markt nutzen und einen Vertreter im Land benennen. Anschließend müssen sie die erforderlichen Unterlagen einreichen, darunter:
- FDA 510(k)/Premarket Approval Letter/CE.
- Zertifikat für ausländische Regierungen (CFG) / Freiverkaufszertifikat (CFS).
- ISO 13485 oder eine andere anerkannte Zertifizierung für gute Herstellungspraktiken (GMPs).
- Validierung und Zertifizierung durch das israelische Normungsinstitut (falls erforderlich).
Prozessablauf
Lebenszyklusmanagement von Medizinprodukten nach der Zulassung
Freyr bietet ausländischen Herstellern umfassende Unterstützung bei der Verwaltung des gesamten Lebenszyklus von Medizinprodukten in Israel, einschließlich der Aktivitäten nach der Zulassung:
- Verwaltung von Änderungen nach der Zulassung, die Anpassungen an bestehenden Medizinproduktezulassungen umfasst, zum Beispiel die Einführung neuer Varianten, Zubehörteile und Anwendungsgebiete.
- Aufrechterhaltung der ISO 13485:2016 und der CE-Zertifizierung.
- Verlängerung von Lizenzen.
- Als Vermittler zwischen der Benannten Stelle (NB) und dem Hersteller fungieren.
Die Bewältigung der Feinheiten von Zulassungsbehörden und die Einhaltung mehrerer Regelwerke für Gerätegenehmigungen kann eine Herausforderung sein. Die Einholung von Genehmigungen aus verschiedenen GHTF-Ländern und die Einhaltung bundesstaatlicher Vorschriften erfordern fundiertes regulatorisches Wissen. Für Markteinsteiger, die diesen Komplexitäten ohne einen etablierten regulatorischen Partner gegenüberstehen, bietet Freyr End-to-End-Regulierungsdienstleistungen an, die den Genehmigungsprozess für Medizinprodukte in Israel optimieren.
Expertise bei der Medizinprodukte-Registrierung in Ägypten
- Israelische Medizinprodukteklassifizierung.
- Israelischer Registrierungsinhaber (IRH).
- Israelische Medizinproduktregistrierung.
- ISO 14971:2019 Beratung zum Risikomanagement.
- ISO 13485:2016 Konformität.
- Überprüfung, Zusammenstellung und Einreichung von Design-Dossiers.
- Registrierung von Medizinprodukten über ein Online-Registrierungssystem.
- Bericht zur regulatorischen Strategie für Medizinprodukte.
- Unterstützung bei Tests – Biokompatibilität, elektrische Sicherheit, mechanische Eigenschaften und Leistung.
- Unterstützung bei der Einhaltung der Kennzeichnungsvorschriften.
- GMP-Unterstützung.
- Unterstützung bei der Post-market Surveillance (PMS).
