

Übersicht zur Registrierung von Medizinprodukten in Neuseeland
Medizinprodukte in Neuseeland werden von der New Zealand Medicines and Medical Devices Safety Authority (Medsafe) gemäß den Medicines Regulations 1984, dem Medicines Act 1981 und den Medicines (Database of Medical Devices) Regulations 2003 reguliert. Eine Genehmigung vor der Markteinführung ist zwar nicht erforderlich, jedoch müssen die Produkte innerhalb von 30 Tagen nach der kommerziellen Einführung in der Datenbank des Electronic Web Assisted Notification of Devices Systems (WAND) gelistet werden. Dokumente zum Nachweis der Sicherheit und Wirksamkeit, wie beispielsweise eine Zertifizierung von anerkannten Stellen wie einer EU-Benannten Stelle oder Health Canada, können von Medsafe angefordert werden.
Das Team von Freyr aus Experten für die Regulierung von Medizinprodukten verfügt über beträchtliche Erfahrung darin, Medizinprodukteunternehmen durch den Medsafe-Registrierungsprozess für Medizinprodukte in Neuseeland zu begleiten.
![]()
Aufsichtsbehörde: Sicherheitsbehörde für Medizinprodukte (Medsafe)![]()
Verordnung:Die Arzneimittel (Datenbank für Medizinprodukte) Verordnung, 2003
Arzneimittelgesetz 1981
Arzneimittelverordnung 1984![]()
Regulatorischer Weg: Elektronisches webgestütztes System zur Meldung von Medizinprodukten (WAND)![]()
Bevollmächtigter Vertreter: Sponsor für Medizinprodukte![]()
QMS-Anforderung: ISO 13485:2016-Zertifizierung![]()
Bewertung technischer Daten: Sicherheitsbehörde für Medizinprodukte (Medsafe)![]()
Gültigkeit der Lizenz: Die Geräteeinträge in Neuseeland laufen nicht ab. Geräte, die als große Gefahr für die Öffentlichkeit eingestuft werden, können vom Markt genommen werden.![]()
Kennzeichnungspflichten: Vorschrift 12(4) der Arzneimittelverordnung 1984 und GHTF/SG1/N43:2005![]()
Einreichungsformat: Elektronisches webgestütztes System zur Meldung von Medizinprodukten (WAND)![]()
Sprache: Englisch
Klassifizierung von Medizinprodukten in Neuseeland
Medizinprodukte in Neuseeland werden gemäß den Kriterien des International Medical Device Regulators Forum (IMDRF) nach Risiko in die Klassen I, IIa, IIb, III und AIMD eingeteilt. Diese Klassifizierung beeinflusst das erforderliche Ausmaß der regulatorischen Kontrolle. Die Klassifizierung basiert auf Merkmalen wie dem Verwendungszweck des Geräts, der Dauer des Körperkontakts, der Invasivität und ob es aktiv oder inaktiv ist. Geräte höherer Klassen unterliegen einer strengeren regulatorischen Aufsicht. Medsafe ist die Regulierungsbehörde in Neuseeland, die diese Klassifizierungen und Vorschriften überwacht.
| Medsafe-Klassifizierung von Medizinprodukten außerhalb der IVD-Klasse | Risiko |
|---|---|
| Klasse I Basis | Geringes Risiko |
| Klasse I messend | Geringes Risiko |
| Klasse I steril | Geringes Risiko |
| Klasse IIa | Niedriges bis mittleres Risiko |
| Klasse IIb | Mittleres bis hohes Risiko |
| Klasse III & Aktive implantierbare Medizinprodukte (AIMD) | Hohes Risiko |
| Medsafe IVD-Klassifizierung | Risiko |
|---|---|
| Seit Juli 2014 erkennt Medsafe kein Risikoklassifizierungssystem für IVDs an. Alle bei WAND gemeldeten IVDs müssen den Risikoklassifizierungscode des IVD verwenden. Der Generaldirektor für Gesundheit genehmigte die Ausnahme für IVDs gemäß Anhang 1, Absatz (i) der Medicines (Database of Medical Devices) Regulations 2003. Lieferanten von IVDs können ihre Produkte jedoch freiwillig in der Datenbank melden. | |
Bevollmächtigter/Sponsor für Medizinprodukte
Der Bevollmächtigte wird als Sponsor bezeichnet und fungiert als Vermittler zwischen dem Hersteller und Medsafe. Sponsoren sind die regulatorischen Vertreter für Produkte, die in Neuseeland vermarktet werden. Sie reichen WAND-Anträge ein und sind die primäre Kontaktstelle zwischen dem Hersteller und Medsafe für alle produktbezogenen Angelegenheiten. Darüber hinaus macht Medsafe den Sponsor für die Vigilanz-Aktivitäten verantwortlich.
Registrierung von Medizinprodukten in Neuseeland
Das Verfahren zur Registrierung von Medizinprodukten und zur WAND-Listung in Neuseeland variiert je nach Geräteklasse.
Geräte der Klasse I – Für nicht-sterile, nicht-messende Geräte der Klasse I ist eine Konformitätserklärung des Herstellers erforderlich; diese wird jedoch selten bei einer Regulierungsbehörde eingereicht. Stattdessen muss der Sponsor (oder Lieferant) die Gerätedetails im Rahmen des Medsafe-Benachrichtigungsprozesses in die WAND-Datenbank (Web Assisted Notification of Devices) eingeben.
Geräte anderer Klassen
In Neuseeland sind Sponsoren oder Lieferanten dafür verantwortlich, dass Medizinprodukte Standards wie ISO 13485:2016 erfüllen. Eine direkte Einreichung einer Konformitätserklärung, einer QMS-Zertifizierung oder eines Herstellungsnachweises bei Medsafe ist normalerweise nicht erforderlich. Das Aufbewahren dieser Dokumentation ist jedoch entscheidend, um die Konformität auf Anfrage nachweisen zu können.
Medsafe priorisiert die Überwachung nach dem Inverkehrbringen (Post-Market Surveillance) gegenüber einer detaillierten Genehmigung vor dem Inverkehrbringen für Medizinprodukte. Während Audits während der Notifizierungsphase nicht routinemäßig durchgeführt werden, kann Medsafe diese für Geräte mit höherem Risiko oder nach Vigilanzaktivitäten und Berichten über unerwünschte Ereignisse einleiten, um die fortgesetzte Sicherheit und Konformität zu gewährleisten.
Sobald ein Produkt über die WAND-Datenbank notifiziert wurde, kann es in Neuseeland vermarktet werden, vorausgesetzt, der Lieferant erfüllt stets die Vorschriften von Medsafe. Dies erfordert eine fortlaufende Einhaltung der Vorschriften, insbesondere der Standards für die Überwachung nach dem Inverkehrbringen und die Meldung von Vorfällen. Die Medizinprodukteexperten von Freyr unterstützen Dienstleistungen im Zusammenhang mit der Navigation durch diese regulatorischen Anforderungen und stellen sicher, dass Unternehmen die Konformität während des gesamten Produktlebenszyklus aufrechterhalten.
Prozessablauf
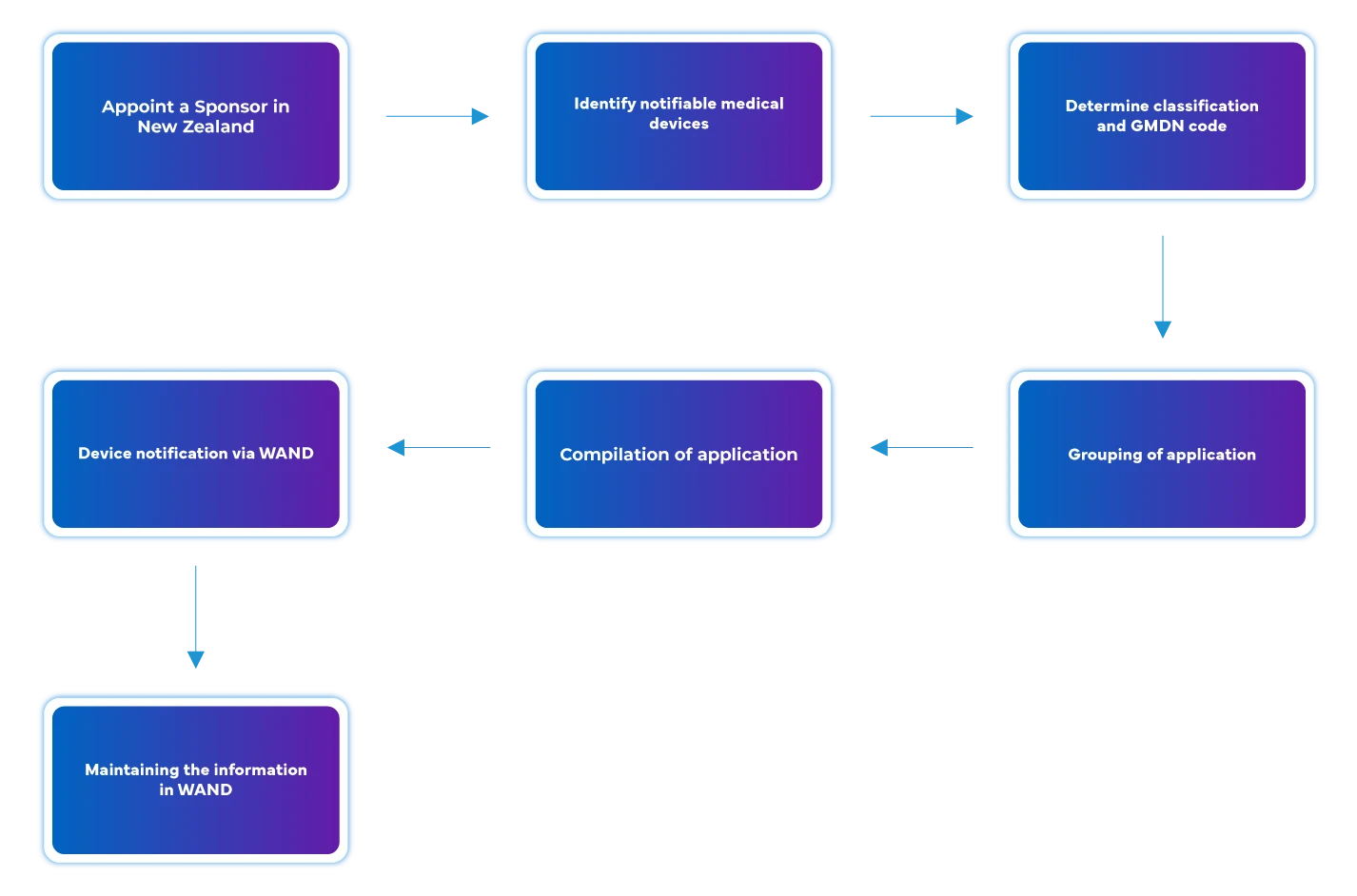
Lebenszyklusmanagement von Medizinprodukten nach der Zulassung
Freyr unterstützt ausländische Hersteller beim End-to-End Lebenszyklusmanagement von Medizinprodukten, einschließlich Aktivitäten nach der Zulassung, durch Benachrichtigung der neuseeländischen Behörden über WAND, wie zum Beispiel –
- Änderungsmanagement nach der Zulassung – Änderungen an bestehenden Zulassungen von Medizinprodukten, wie zum Beispiel die Hinzufügung neuer Varianten, Zubehörteile oder neuer Anwendungsindikationen.
- Aufrechterhaltung von Zulassungen und Registrierungen.
Mit einem Team von Regulierungsexperten bietet Freyr Herstellern umfassende Unterstützung, um die für die Marktzulassung erforderlichen Qualitäts- und Sicherheitsstandards einzuhalten. Die Spezialisten für regulatorische Intelligenz des Unternehmens überwachen sorgfältig Aktualisierungen der Vorschriften und stellen sicher, dass Kunden gut über die notwendigen Maßnahmen zur Aufrechterhaltung der Konformität ihrer Produkte mit den aktuellen Standards informiert sind.
Zusammenfassung
| Risiko | Geräteklasse | QMS-Audit | Regulatorischer Weg | Medsafe-Fristen | Gültigkeit der Registrierung (Jahre) |
|---|---|---|---|---|---|
| Geringes Risiko | Klasse I Basis | ISO 13485:2016 Konformität Hinweis: Medsafe verlangt keine QMS-Audits, empfiehlt jedoch dringend, ISO 13485:2016 für Qualität und Sicherheit zu befolgen. Medsafe ist befugt, QMS-Audits für jede Geräteklasse durchzuführen, wenn Sicherheits- oder Qualitätsbedenken auftreten. | WAND-Listung (Benachrichtigung) | 1 Woche |
Keine Ablaufdaten |
| Geringes Risiko | Klasse I messend | WAND-Listung (Benachrichtigung) | |||
| Geringes Risiko | Klasse I steril | WAND-Listung (Benachrichtigung) | |||
| Niedriges bis mittleres Risiko | Klasse IIa | WAND-Listung (Benachrichtigung) | |||
| Mittleres bis hohes Risiko | Klasse IIb | WAND-Listung (Benachrichtigung) | |||
| Hohes Risiko | Klasse III | WAND-Listung (Benachrichtigung) |
Hinweis: Gemäß der aktuellen Gesetzgebung laufen Geräteeintragungen in Neuseeland nicht ab. Geräte, die als unannehmbares Risiko für die Öffentlichkeit eingestuft werden, können jedoch vom Markt genommen werden. Die aktuelle Gesetzgebung könnte jedoch bis 2026/2027 überarbeitet werden.
Freyr Expertise
- Umfassende Unterstützung bei der Registrierung von Medizinprodukten.
- LR-Unterstützung
- WAND-Eintragung
- Kennzeichnungsunterstützung
- Änderungsmanagement nach der Zulassung
- Lizenzübertragung
- Einreichungs- und Vermittlungsdienste mit WAND