4 Minuten lesen
Die Meldung von Medizinprodukten (Medical Device Reporting, MDR) ist ein Instrument zur Überwachung nach dem Inverkehrbringen, das die Food and Drug Administration (FDA) einsetzt, um die Leistung von Produkten zu überwachen, potenzielle sicherheitsrelevante Probleme im Zusammenhang mit Produkten zu erkennen und zur Bewertung des Nutzen-Risiko-Verhältnisses von Produkten beizutragen. Der Zweck von MDR besteht darin, unerwünschte Ereignisse im Zusammenhang mit Produkten rechtzeitig zu erkennen und zu beheben. Es ermöglicht Ärzten, Gesundheitseinrichtungen, Herstellern und Verbrauchern, freiwillige Meldungen zu machen, um die Sicherheit und Wirksamkeit des Produkts nach dem Inverkehrbringen zu verstehen.
Die MDR gilt für alle Klassen von Medizinprodukten, die entweder in den Vereinigten Staaten von Amerika (USA) hergestellt oder in die USA eingeführt werden. Hersteller von Medizinprodukten, die ihre Produkte in den USA vermarkten wollen, müssen die MDR einhalten, andernfalls kann es zu finanziellen Sanktionen kommen. Sie gilt in den USA einschließlich eines ausländischen Ereignisses, d. h. sie ist auf legal in den USA in Verkehr gebrachte Medizinprodukte anwendbar, die sowohl in den USA als auch im Ausland hergestellt wurden. Darüber hinaus gibt es verschiedene Fälle der Anwendbarkeit einer MDR, wie z.B.:
- wenn ein Produkt in den USA hergestellt, vor Ort und auf anderen Märkten vertrieben wird
- wenn ein Produkt in den USA hergestellt, aber auf anderen Märkten vertrieben wird
- wenn ein Gerät im Ausland hergestellt und in die USA und andere Märkte geliefert wird
- wenn ein Produkt im Ausland hergestellt und vor Ort vertrieben wird und
- wenn ein Gerät in den USA untersucht wird
MDR und der Ablauf des Meldeprozesses
Die MDR-Verordnung enthält zahlreiche verbindliche Anforderungen für Hersteller, Importeure und Einrichtungen, die Medizinprodukte verwenden, bestimmte produktbezogene unerwünschte Ereignisse und Produktprobleme an die FDA zu melden. Das unten aufgeführte Prozess-Flussdiagramm beschreibt den Meldeprozess Schritt für Schritt.
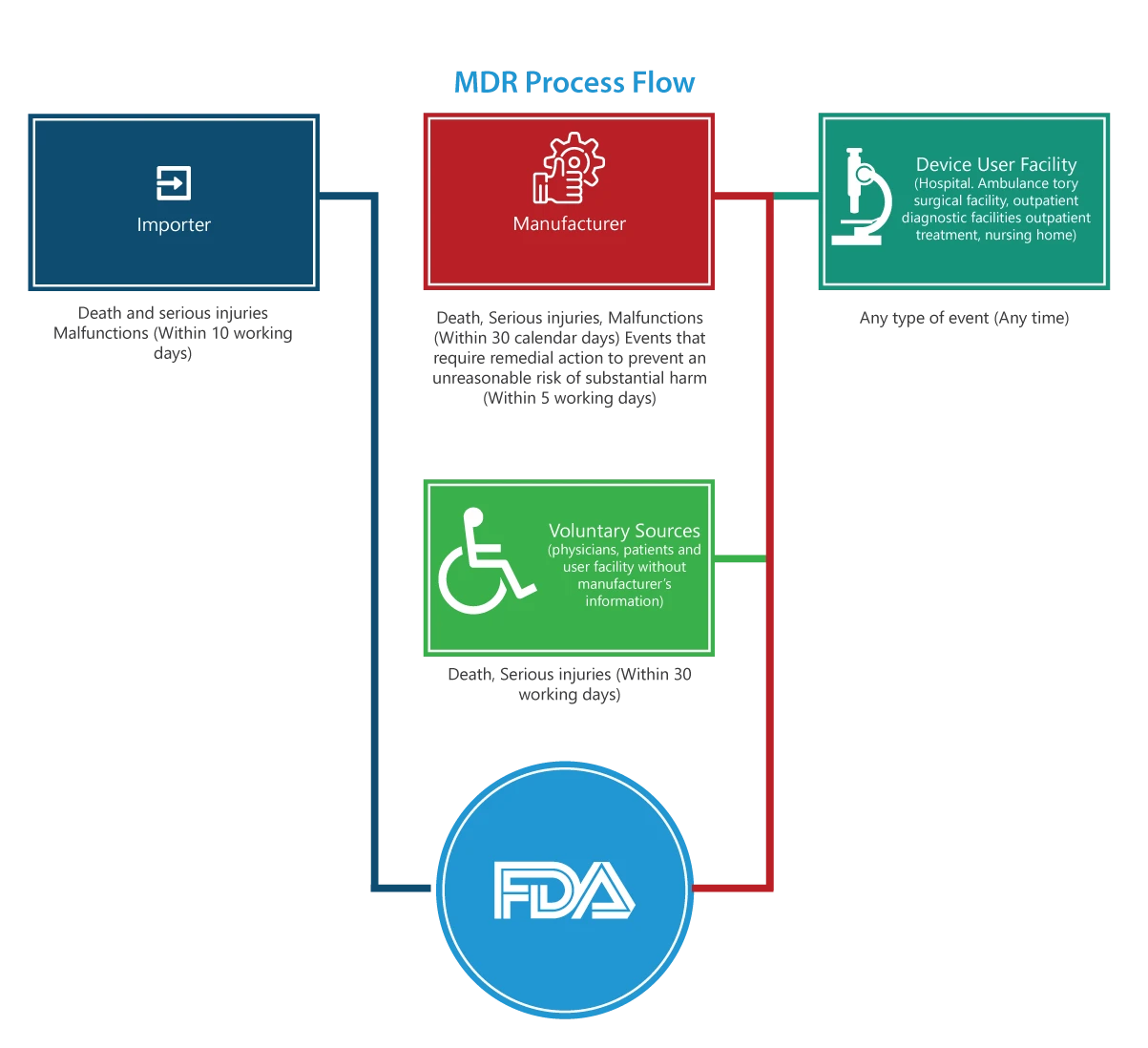
Für wen gilt sie?
Importeure
Meldungen über Todesfälle, schwere Verletzungen und Fehlfunktionen müssen innerhalb von 30 Arbeitstagen an die FDA den Hersteller übermittelt werden. Wenn die Fehlfunktion an anderer Stelle zu Verletzungen oder Todesfällen führen kann, müssen die Importeure die Fehlfunktion dem Hersteller melden.
Hersteller
Meldungen zu einem von FDA festgelegten Ereignis (Todesfälle, schwere Verletzungen und Fehlfunktionen) FDA zu einem Ereignis, das Abhilfemaßnahmen erfordert, um ein unangemessenes Risiko einer erheblichen Gefährdung der öffentlichen Gesundheit zu vermeiden, müssen FDA fünf Arbeitstagen unter Verwendung des Formulars 3500A bei der FDA eingereicht werden.
Einrichtung des Geräteanwenders (Krankenhaus, ambulante chirurgische Einrichtung, Pflegeheim, ambulante Diagnoseeinrichtung oder ambulante Behandlungseinrichtung)
Meldungen müssen spätestens 10 Werktage nach dem Tag, an dem der Hersteller Kenntnis davon erlangt hat, dass ein Produkt eine schwere Verletzung eines Patienten der Einrichtung verursacht oder dazu beigetragen hat oder haben könnte, an den Hersteller des Produkts übermittelt werden. Ist der Hersteller unbekannt, muss die Einrichtung die Meldung an die FDA übermitteln.
Freiwillige Gruppen
Patienten, medizinisches Fachpersonal und Verbraucher, die ein Problem im Zusammenhang mit einem Medizinprodukt feststellen, können dies FDA MedWatch an FDA melden.
eMDR
Die FDA 2015 die elektronische MDR (eMDR) FDA , um kritische Probleme hinsichtlich der Datenqualität und -integrität im Zusammenhang mit der Meldung schwerwiegender Verletzungen im Zusammenhang mit allen Klassen von Medizinprodukten zu identifizieren. eMDR wird als Meldeverfahren bevorzugt.
Hersteller können ihre eMDR über ein Electronic Submissions Gateway (ESG) einreichen. Nach der Einreichung dauert es bis zu 48 Stunden, bis das elektronische Gateway eine Bestätigung sendet. Wenn bei der Einreichung des Berichts ein Fehler auftritt, wird eine Meldung angezeigt, damit die Korrektur(en) vorgenommen werden können.
eMDR - Welchen Nutzen hat es?
eMDR bietet zahlreiche Vorteile gegenüber dem manuellen Meldemechanismus (d. h. MDR). Nachstehend sind einige bemerkenswerte Vorteile aufgeführt, auf die sich Hersteller, Agenturen und Patienten verlassen können:
- Das eMDR-Einreichungstool verbessert die Zusammenarbeit zwischen einem Unternehmen, der Gesundheitsbehörde (FDA) und Patienten.
- eMDR spart Kosten. Durch die Automatisierung werden der Verwaltungsaufwand und die herkömmliche Kommunikation reduziert. Dies trägt zur Beschleunigung des Prozesses bei und fördert eine effektive Meldung von Ereignissen, was zu einer sofortigen Interaktion mit der FDA führt.
- Manuelle Verfahren sind mit viel Papierkram verbunden, können langwierig und schwierig zu verfolgen und zu bearbeiten sein. eMDR-Anträge sind automatisiert und zentralisiert. Aufzeichnungen können leicht abgerufen werden, was viel Zeit bei der Überprüfung spart.
- Mit eMDR können die Beteiligten Fehler bei der Einreichung schnell melden, anstatt manuell und zeitaufwendig mit der FDA zu korrespondieren.
- eMDR fungiert als zentrale Anlaufstelle für die Bearbeitung aller elektronischen Einreichungen in einer hochsicheren Umgebung und ist von Vorteil, da Beschwerden innerhalb der Organisation direkt mit dem MedWatch-Formular verknüpft und in das Gateway FDAintegriert werden können.
eMDR und der Ablauf des Meldeprozesses
Die eMDR-Verordnung enthält verbindliche Anforderungen für Hersteller, Importeure und Einrichtungen, die Medizinprodukte verwenden, bestimmte produktbezogene unerwünschte Ereignisse und Produktprobleme an die FDA zu melden. Das folgende Flussdiagramm beschreibt den Meldeprozess Schritt für Schritt.
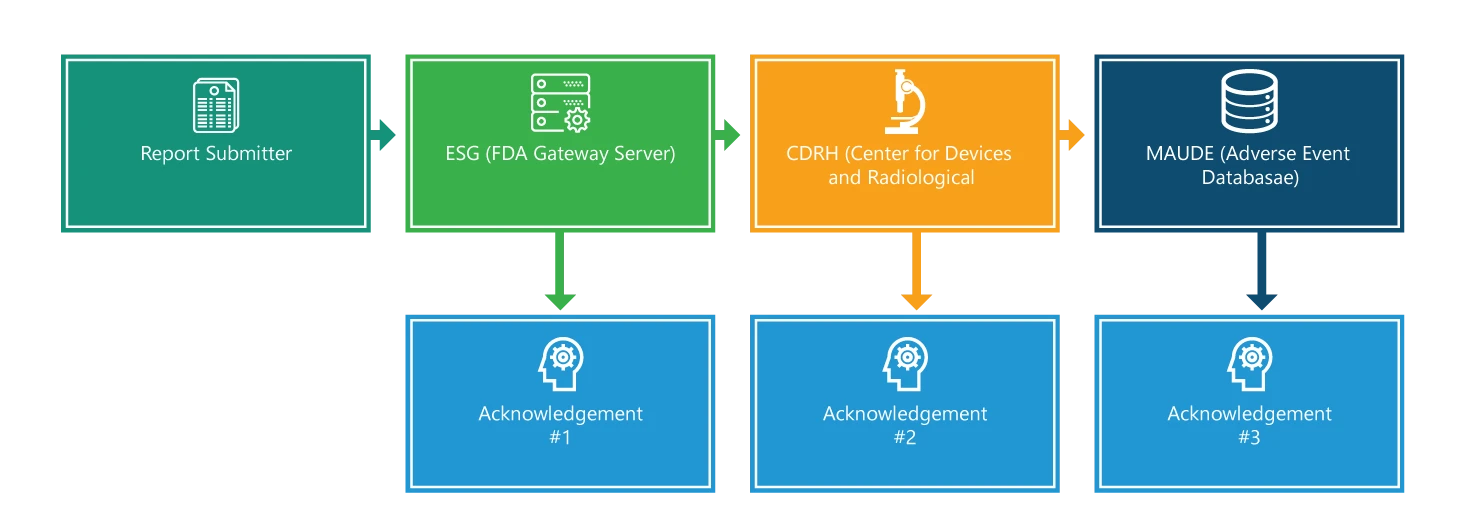
Das Meldeverfahren besteht aus vier Schritten. Mit Ausnahme des ersten Schrittes wird jeder Schritt bestätigt. Darüber hinaus wird jeder Schritt mit zusätzlichen Informationen versehen, die den Prozess erleichtern sollen.
Schritt 1: Einreicher des Berichts
Einreichen eines eMDR. Um einen Antrag einzureichen, benötigen Sie zunächst eine elektronische Signatur und müssen sicherstellen, dass die Dateinamen nur einen Punkt enthalten, der zur Angabe der Dateiendung dient (z. B. 555.xml 555.pdf). Die Übermittlungs- und Bearbeitungszeit hängt jedoch von der Gesamtgröße Ihres Antrags ab. Größere Anträge benötigen mehr Zeit für die Übermittlung und Bearbeitung.
Schritt 2: Elektronisches Einreichungsportal (ESG)
Wenn Ihre Einreichung bei der ESG eingeht, sollten Sie umgehend eine Bestätigung Nr. 1 erhalten, es sei denn, die ESG wegen Wartungsarbeiten nicht erreichbar. Sie müssen den Status Ihrer MDR auf der ESG überprüfen.
Schritt 3: CRDH
eMDR wird automatisch vom ESG das Center for Devices and Radiological Health (CDRH) weitergeleitet. Sobald die Weiterleitung erfolgt ist, sollten Sie wie in Schritt 2 eine Bestätigung erhalten, d. h. Nr. 2.
Schritt 4: Hersteller- und Nutzererfahrung mit Geräten (MAUDE)
Wenn das CDRH die Meldung validiert und in der Datenbank für unerwünschte Ereignisse (MAUDE) aktualisiert hat, wird erwartet, dass der Einsender eine Bestätigungsmeldung Nr. 3 erhält. Es ist zu beachten, dass alle Fehler, die während der Validierung und des Ladens auftreten, aufgezeichnet werden.
Die Meldung von Medizinprodukten (Medical Device Reporting, MDR) ist ein wichtiger Prozess, der hilft, Leben zu retten und Patienten vor unnötigen Risiken zu schützen. Er stellt sicher, dass alle an der Patientenversorgung beteiligten Parteien verantwortungsbewusst und aufmerksam mit den Produkten umgehen.
eMDR erleichtert die Berichterstattung, aber die Dokumentation und Nachverfolgung können ressourcenintensiv sein. Machen Sie es gleich beim ersten Mal richtig – sprechen Sie mit us sales@freyrsolutions.com.