
3 min ler
A partir de 31 de janeiro de 2022, o novo regulamento farmacêutico da União Europeia (UE) para o Regulamento de Ensaios Clínicos (CTR) tornou-se obrigatório, revogando a Diretiva de Ensaios Clínicos 2001/20/CE. O regulamento harmoniza os protocolos de avaliação e supervisão de ensaios clínicos em toda a UE. As diretrizes foram revistas para promover uma abordagem uniforme à investigação clínica, salientando simultaneamente a segurança dos participantes nos ensaios clínicos e uma maior divulgação pública.
O regulamento estabelece um novo sistema de avaliação em duas partes para todos os ensaios clínicos da UE. A Parte I consiste numa avaliação científica da documentação de base do ensaio clínico e a Parte II consiste numa avaliação ética da documentação a nível nacional. Na sequência desta avaliação em duas partes, cada Estado-Membro tomará uma decisão unificada sobre o ensaio e notificará o promotor através do Sistema de Informação sobre Ensaios Clínicos.
Prazos de transição para novos candidatos
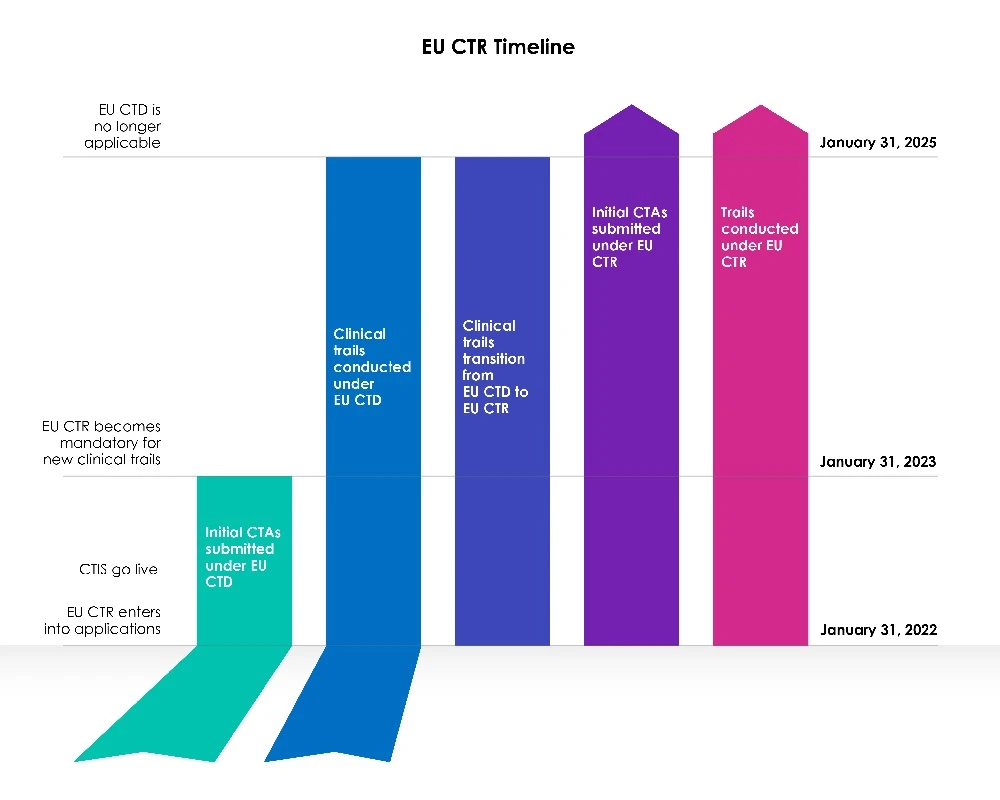
Uma fase de transição de três (03) anos teve início com a data CTIS da UE.
Ano 1 (31 de janeiro de 2022 a 30 de janeiro de 2023):
A Diretiva 2001/20/CE da União Europeia (UE) relativa aos ensaios clínicos (EU-CTD) rege os ensaios clínicos na UE desde 2004. Ela buscou padronizar as regras e melhorou significativamente a segurança dos pacientes em ensaios clínicos. No entanto, na prática, ela criou consequências indesejadas. Durante o primeiro ano, após CTIS , os patrocinadores podiam escolher entre solicitar uma nova submissão de ensaio clínico submissão CTA) ao abrigo do Sistema de Informação sobre Ensaios Clínicos (CTIS) da Diretiva relativa aos ensaios clínicos (CTD: Diretiva 2001/20/CE) ou utilizar CTIS conformidade com a legislação em vigor, o Regulamento (UE) n.º 536/2014 relativo aos ensaios clínicos.
Ambas as ideias eram viáveis e os patrocinadores tiveram a oportunidade de escolher a legislação a adotar.
Os membros estavam prontos para utilizar o Sistema de Informação sobre Ensaios Clínicos (CTIS) e aceitaram os pedidos ao abrigo da nova legislação, o Regulamento relativo aos ensaios clínicos (EU CTR), no primeiro dia de CTIS .
Anos 2 e 3 (31 de janeiro de 2023 a 31 de janeiro de 2025):
A partir de 31 de janeiro de 2023, todos os novos pedidos de CT devem ser apresentados através do CTIS a nova legislação (CTR).
Não é permitido apresentar novos pedidos de CT no EudraCT ao abrigo da Diretiva relativa aos ensaios clínicos (CTD). A Diretiva relativa aos ensaios clínicos da UE já não permite a admissão de novos Member States 31 de janeiro de 2023. Os ensaios realizados ao abrigo da CTD devem primeiro ser transferidos, após o que submissão ser apresentado um submissão adicional relativo ao Estado-Membro em causa através CTIS EU CTIS.
Para candidatos existentes
Os pedidos de autorização de ensaios clínicos apresentados antes de 30 de janeiro de 2023, ao abrigo da antiga legislação (CTD), utilizando a EudraCT, serão autorizados a decorrer até à conclusão ao abrigo dessa diretiva (CTD: Diretiva 2001/20/CE), até 30 de janeiro de 2025. Os procedimentos manter-se-ão inalterados e os promotores poderão apresentar alterações significativas e avisos de fim de ensaio, tal como exigido pelo regulamento. A base EudraCT permanecerá ativa durante o período de transição para permitir a continuação destes ensaios.
É, no entanto, importante notar que os pedidos de transição podem ser apresentados em qualquer altura durante o período de transição de três (03) anos, e os promotores são encorajados a concluir o processo suficientemente cedo no período de transição para assegurar a continuidade do ensaio clínico na UE para além de 30 de janeiro de 2025, tendo em conta os feriados legais e as duas (02) semanas de paragem do relógio de inverno.
Ensaios não transferíveis
- Os ensaios que já foram concluídos ou que serão concluídos pouco antes do final do períodoEEA não devem ser transferidos.
- Se uma notificação de fim de ensaio tiver sido concluída em todos os paísesEEA , mas o fim global do ensaio ainda não tiver sido notificado, o estudo não deve ser transferido. De acordo com a diretiva, o fim global do ensaio e os resultados resumidos do ensaio devem ser publicados através do EudraCT.
- Os ensaios que tiveram início antes da implementação da Diretiva 2001/20/CE não beneficiam desse procedimento de transição. Se forem intervencionais e tiverem de continuar a funcionar após o término da fase de transição do CTR, submissão ser emitida uma nova submissão CT ao abrigo do CTR.
- Os ensaios pediátricos realizados fora daEEA aos quais foi atribuído um número EudraCT, também não devem ser convertidos.
- Os ensaios que estiverem suspensos após o término do período de transição não poderão ser transferidos. Reiniciar o ensaio nessas circunstâncias exigiria o submissão de um novo submissão CTR.
CTIS da UE CTIS atualizações técnicas EMA da EMA para melhorar as suas características e funcionalidades. Quando são feitas alterações significativas no CTIS, a EMA notas de lançamento descrevendo o que foi alterado no sistema. As atualizações podem incluir melhorias nas características e funcionalidades existentes, a adição de novas características e melhorias funcionais e técnicas. Um parceiro regulatório experiente pode lidar com potenciais desafios e ajudar os patrocinadores na transição de ensaios clínicos existentes e futuros, como parte das estratégias de desenvolvimento clínico. Clique aqui para saber mais sobre o CTIS a experiência Freyrna área: https://regulatoryaffairs.freyrsolutions.com/clinical-trial-applications-ctas.









