
5 min. de leitura
Medical Device Single Audit Program MDSAP) permite que uma Organização de Auditoria (AO) reconhecida realize uma única auditoria do Quality Management System (QMS) de um fabricante de dispositivos médicos. Ele fornece requisitos regulatórios relevantes para cinco países, ou seja, Brasil (ANVISA), EUA (FDA), Japão (PMDA), Canadá (Health Canada) e Austrália (TGA). Além das autoridades regulatórias participantes, vários outros parceiros internacionais (os observadores oficiais e membros afiliados) estão envolvidos no MDSAP.
MDSAP é exigida pela Health Canada dispositivos das classes II, III e IV, mas é voluntária para os outros quatro países. Ela promoveu a transparência e o alinhamento regulatório entre as autoridades participantes e minimizou a necessidade de múltiplas auditorias, economizando tempo e recursos dos fabricantes de dispositivos médicos. Para lhe dar uma melhor perspectiva sobre o MDSAP , tentamos responder aqui às quinze (15) perguntas mais frequentes.
- Por que foi MDSAP foi desenvolvido quando existe uma ISO 13485 globalmente aceita?
MDSAP desenvolvido para reduzir o peso das auditorias regulatórias para fabricantes de dispositivos médicos e promover um maior alinhamento das abordagens regulatórias e requisitos técnicos com base em normas internacionais e melhores práticas. O seu foco é trazer consistência, previsibilidade e transparência aos programas regulatórios, padronizando procedimentos e práticas de reguladores e organizações de auditoria terceirizadas.
A auditoria baseia-se nos requisitos do SGQ da norma ISO 13485 nos requisitos regulamentares do país participante onde os dispositivos médicos serão comercializados.
- Quais são os critérios de elegibilidade para se submeter a uma MDSAP ?
Qualquer fabricante de dispositivos médicos que pretenda comercializar os seus dispositivos nos países participantes pode submeter-se a uma MDSAP . No entanto, cada autoridade reguladora pode estabelecer critérios de exclusão para determinadas condições, se necessário.
Por exemplo, no Japão, as excepções à elegibilidade são:
- Um local de fabrico registado (RMS) que fabrica dispositivos médicos feitos de tecidos humanos/animais
- Um SGR que fabrica DIV radioactivos, e
- Estabelecimento de um Autorização de Introdução no Mercado (MAH)
- A MDSAP inclui produtos combinados?
Os dispositivos médicos que incluem medicamentos (substâncias medicinais) ou produtos biológicos (por exemplo, materiais de origem animal que foram tornados inviáveis, ou tecidos, células ou substâncias de origem microbiana ou recombinante, sangue humano ou extratos de sangue humano ou produtos sanguíneos, etc.) são considerados produtos combinados e podem ser incluídos no âmbito de uma MDSAP .
No entanto, devido às diferenças na forma como esses produtos são regulamentados nas jurisdições das autoridades reguladoras participantes, os relatórios MDSAP e os documentos de certificação podem não ser considerados uma alternativa aos requisitos de inspeção e avaliação em algumas jurisdições.
Austrália - Os produtos combinados estão sujeitos a uma inspeção externa da TGA (Agência Australiana de Medicamentos) ao abrigo da Avaliação de Conformidade Australiana. No entanto, uma MDSAP eficaz pode reduzir as inspeções para estes dispositivos.
Brasil, Japão - Os produtos combinados considerados dispositivos médicos estão incluídos no MDSAP, uma vez que não existem requisitos específicos relativos ao SGQ.
Canadá - MDSAP abrange os requisitos do SGQ para produtos combinados considerados dispositivos médicos.
EUA - MDSAP não são consideradas alternativas às FDA para produtos combinados.
- Posso selecionar o país abrangido pela MDSAP ?
Sim, a auditoria é realizada de acordo com o escopo declarado na submissão serviços de certificação. Espera-se que os fabricantes de dispositivos médicos estejam em conformidade com as regulamentações apenas nas jurisdições onde os seus produtos serão comercializados.
- Sou um fabricante de dispositivos médicos dos US e pretendo comercializar o meu dispositivo apenas no Japão. Estou prestes a passar por uma MDSAP . Preciso de cumprir os requisitos de outros países também?
Não, os fabricantes de dispositivos médicos só precisam estar em conformidade com ISO 13485 e regulamentos ISO 13485 nas jurisdições onde os seus produtos serão comercializados.
- A minha Organização de Auditoria (AO) e o Organismo Notificado Europeu são o mesmo. Posso ser auditado por ambos ao mesmo tempo?
Se o seu AO e o Organismo Notificado Europeu forem os mesmos, a avaliação de conformidade pode ser realizada após a realização da MDSAP , e não simultaneamente. Os Organismos Notificados Europeus são observadores do MDSAP, e a avaliação de conformidade é realizada de acordo com o EU MDR . Para MDSAP, a avaliação é realizada de acordo com os requisitos da ISO 13485 os requisitos regulamentares dos países participantes no âmbito.
- Qual é a diferença entre as avaliações de Fase I e II?
O processo de auditoria MDSAP envolve duas etapas. A auditoria inicial, também chamada de auditoria de certificação inicial, consiste nas auditorias da Etapa I e da Etapa II.
A auditoria da Fase I inclui a revisão da documentação e a avaliação do grau de preparação do fabricante de dispositivos médicos para se submeter a uma auditoria da Fase II.
A auditoria de fase II é realizada para verificar se todos os requisitos aplicáveis da ISO 13485 outros requisitos regulamentares da autoridade reguladora em vigor estão implementados.
- Quantos auditores posso esperar para uma MDSAP ?
A Determinação do Tempo de Auditoria especifica como determinar a duração da auditoria no local em dias-homem. AO decide quantos auditores irão compor a equipa de auditoria. Por exemplo, uma auditoria de (06) dias-homem pode ser concluída em três (03) dias por uma equipa de dois (02) auditores.
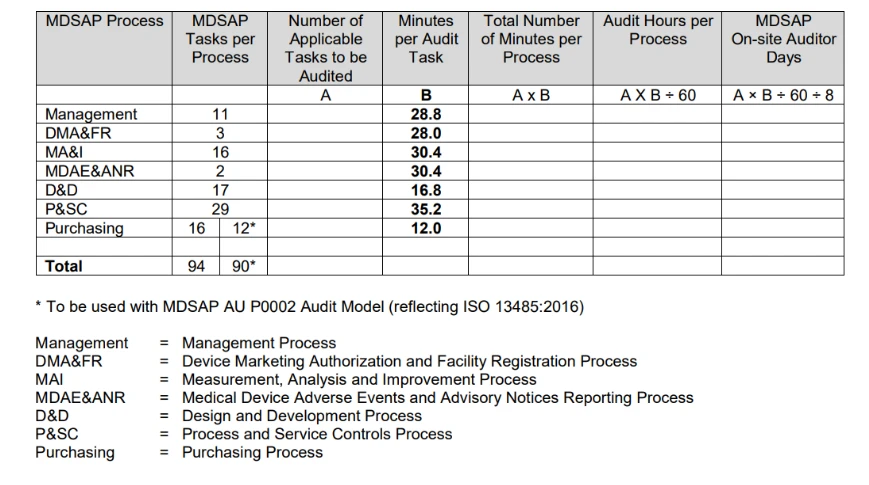
- Como é que a MDSAP é cronometrada?
O Procedimento de Determinação do Tempo de Auditoria, emitido pela FDA, resume o processo para determinar a duração do cálculo da auditoria na tabela a seguir.
O cálculo da duração da auditoria baseia-se principalmente no número de tarefas de auditoria aplicáveis associadas ao tipo de auditoria a realizar e às actividades específicas da organização a auditar.
Para obter informações detalhadas sobre o mesmo, pode consultar o MDSAP .
- Existe algum guia ou lista de verificação que eu possa acessar para garantir a conformidade com uma MDSAP ?
Sim, pode aceder ao documento MDSAP Approach (Abordagem de Auditoria MDSAP). Trata-se de um guia bem organizado, publicado pela USFDA faz referência a secções específicas da ISO 13485:2016 e regulamentos relevantes publicados pela TGA da Austrália, ANVISA do Brasil, Health Canada do Canadá,PMDA do Japão eFDA US .
- Qual é o papel de um observador numa MDSAP ?
MDSAP é uma autoridade reguladora que tem permissão para participar de reuniões, avaliações e outras atividades, mas não utiliza os MDSAP . Os observadores são representados no Conselho da Autoridade MDSAP (RAC) por um gestor de nível sênior.
- Quais são os próximos passos a dar se tiver recebido uma classificação igual ou superior a 4?
O sistema de classificação é atribuído às não-conformidades observadas durante a auditoria pela AO. Uma classificação de 4 ou 5 indica um risco elevado de intervenção. Deve apresentar um plano de correção para cada não conformidade registada no prazo de 15 dias de calendário a contar da data de emissão do relatório de não conformidade. O plano de correção deve incluir os resultados da investigação da não-conformidade, as suas causas e as acções corretivas planeadas para evitar a sua recorrência. As provas da aplicação do plano/ação de correção devem ser apresentadas no prazo de trinta (30) dias de calendário a contar da data de conclusão da auditoria.
- Existe alguma diferença no processo de abordagem da auditoria por um auditor interno em relação a um auditor operacional?
MDSAP uma abordagem de processo. É provável que a AO analise as ligações e os fios condutores, enquanto um auditor interno pode analisar mais detalhadamente um aspeto funcional de cada vez. Portanto, a AO pode encontrar uma não conformidade numa área funcional e procurar respostas numa área funcional diferente. No entanto, seguir a abordagem de processo pode ser perturbador durante uma auditoria interna.
- Posso recorrer ao AO se conseguir provar que uma não conformidade registada não é válida?
A AO tem um processo de recurso ou disputa, que pode ser utilizado se for possível demonstrar que uma não conformidade registada é inválida. No entanto, as classificações atribuídas a não conformidades não podem ser alteradas devido a acções corretivas. Só podem ser alteradas com base em provas que demonstrem que não eram válidas.
- Qual é o prazo de validade do MDSAP ?
Os fabricantes de dispositivos médicos certificados pelo MDSAP serão auditados anualmente, de acordo com um ciclo de certificação de três anos. A auditoria inicial é uma auditoria completa do SGQ do fabricante de dispositivos médicos. É seguida por auditorias de vigilância realizadas anualmente durante dois (02) anos consecutivos. O ciclo recomeça com uma auditoria de recertificação no terceiro ano.
Para saber mais sobre MDSAP nossos MDSAP , entre em contacto com Freyr hoje mesmo para agendar uma chamada com os nossos especialistas.









