Was ist regulatorische Kennzeichnung?
Zuletzt aktualisiert am: August 2024
Die regulatorische Kennzeichnung von Pharmazeutika umfasst die Erstellung, Überprüfung und Verwaltung wichtiger Dokumente, die wesentliche Produktinformationen an Interessengruppen kommunizieren und die Einhaltung globaler regulatorischer Standards gewährleisten. Zu den Kernkomponenten gehören das Core Data Sheet (CDS) und das Company Core Data Sheet (CCDS), die aus Quellen wie Prüfarztbroschüren und Post-Marketing-Daten abgeleitet werden. Dieser Prozess ist entscheidend für die Übermittlung von Sicherheits- und Wirksamkeitsinformationen auf länderspezifischen Etiketten und die Abstimmung mit den Anforderungen der Gesundheitsbehörden (HA).
Mit einem Fokus auf globale Harmonisierung berücksichtigt die regulatorische Kennzeichnung von Arzneimitteln die sich entwickelnden regulatorischen Anforderungen, einschließlich neuer Produktzulassungen, Einreichungen bei Gesundheitsbehörden (HA), Nachzulassungen und des Lebenszyklusmanagements. Präzision und die Einhaltung sich ändernder Richtlinien sind entscheidend für eine erfolgreiche regulatorische Kennzeichnung von Arzneimitteln, da sie die Marktzulassung, das Sicherheitsprofil und die allgemeine regulatorische Lebensfähigkeit eines Produkts beeinflussen.

Freyr, ein führender Anbieter von End-to-End-Dienstleistungen für die regulatorische Kennzeichnung von Arzneimitteln, verfügt über ein engagiertes Team von über 180 globalen Kennzeichnungsexperten, die sich durch die Erstellung wichtiger Dokumente wie Prüfarztbroschüren (IB), Entwicklungs-Kerndatenblätter und Entwicklungs-Kernsicherheitsinformationen auszeichnen. Der Einsatz von künstlicher Intelligenz erhöht die Genauigkeit und beschleunigt die Implementierung und Überprüfung von Datenblättern. Mit einer optimierten CCDS-Vorlage und präzisionsgesteuerten Prozessen erfüllen die umfassenden Dienstleistungen von Freyr effektiv die dynamischen Anforderungen der pharmazeutischen Industrie und bieten eine unvergleichliche Unterstützung für die Einhaltung von Kennzeichnungsvorschriften und den regulatorischen Erfolg.
Warum ist regulatorische Kennzeichnung in der Pharmabranche wichtig?
- Gewährleistung der Patientensicherheit und Informationskommunikation: Die regulatorische Kennzeichnung ist entscheidend für die Patientensicherheit. Etiketten liefern wichtige Informationen über die Anwendung von Medikamenten, Dosierungen, Nebenwirkungen und Gegenanzeigen. Patienten, verschreibende Ärzte, medizinisches Fachpersonal und Pflegekräfte verlassen sich auf diese Etiketten, um fundierte Entscheidungen zu treffen. Eine klare und genaue Kennzeichnung reduziert das Risiko von Medikationsfehlern, unerwünschten Ereignissen und Missbrauch.
Sie stellt sicher, dass Patienten die richtige Behandlung erhalten und deren korrekte Anwendung verstehen. Darüber hinaus verlangen die Regulierungsbehörden, dass jedes auf dem Markt befindliche pharmazeutische Produkt eine Kennzeichnung aufweisen muss, um Behandlungsinformationen effektiv zu kommunizieren. - Einhaltung von Vorschriften und Risikominderung: Die Einhaltung von Kennzeichnungsvorschriften ist nicht nur eine Formalität, sondern eine gesetzliche Anforderung. Regulierungsbehörden wie die US Food and Drug Administration (FDA), die European Medicines Agency (EMA) und andere schreiben eine genaue und umfassende Kennzeichnung vor. Nichteinhaltung kann zu regulatorischen Strafen, einer Schädigung des Markenrufs und sogar zu vorübergehenden Stilllegungen von Produktionslinien führen. Pharmaunternehmen müssen nachweisen, dass ihre Kennzeichnungsprozesse, -methoden, -tests und -geräte in der Lage sind, durchgängig sichere und wirksame Produkte herzustellen. Eine ordnungsgemäß validierte Kennzeichnung mindert Risiken und gewährleistet die Einhaltung der guten Herstellungspraxis (GMP).
- Marktzugang und globale Harmonisierung: Gut strukturierte Etiketten erleichtern den globalen Marktzugang. Eine konsistente Kennzeichnung über Regionen hinweg optimiert Prozesse, reduziert Redundanzen und stimmt mit harmonisierten Standards überein. Da internationale Aufsichtsbehörden GMP-Validierungsanforderungen, einschließlich der Serialisierung, übernehmen, stehen pharmazeutische Lieferketten vor zunehmender Komplexität. Unternehmen, die der Kennzeichnungskonformität Priorität einräumen, schaffen Vertrauen, erhöhen die Marktakzeptanz und positionieren sich für den Erfolg in einem wettbewerbsintensiven Umfeld.
Was ist der Kennzeichnungsgenehmigungsprozess?
Der Genehmigungsprozess für die Kennzeichnung in der pharmazeutischen Industrie umfasst mehrere Phasen, um sicherzustellen, dass alle arzneimittelbezogenen Informationen für medizinisches Fachpersonal und Patienten korrekt, konform und klar sind. Er beginnt mit der Erstellung des Etiketteninhalts, der Details zu Dosierung, Verabreichung, Sicherheit und Warnhinweisen enthält. Regulatorische und medizinische Teams überprüfen diesen Entwurf dann intern, um sicherzustellen, dass er den lokalen und internationalen regulatorischen Standards entspricht. Nach der Fertigstellung wird das Etikett zur Genehmigung bei den Gesundheitsbehörden eingereicht, wo es einer strengen Prüfung unterzogen wird, um die Einhaltung der Sicherheits- und Wirksamkeitsanforderungen zu überprüfen. Erst nach Erhalt der offiziellen Genehmigung darf das Etikett zur Vermarktung des Arzneimittels verwendet werden.
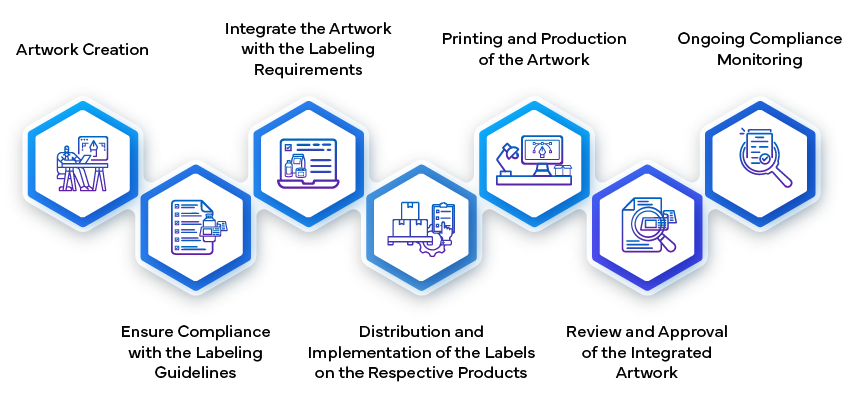
- Erstellung und Einreichung des Etiketts:
- Erste Datenerfassung: Pharmazeutische Unternehmen sammeln alle relevanten Daten für das Arzneimitteletikett. Dies umfasst Informationen zu Wirksamkeit, Sicherheit, Dosierungen, Indikationen, Gegenanzeigen und potenziellen Risiken. Das Etikett dient als wichtiges Kommunikationsmittel für medizinisches Fachpersonal und Patienten.
- Einreichung bei Aufsichtsbehörden: Das Unternehmen reicht die Etikettendaten bei Aufsichtsbehörden wie der US Food and Drug Administration (FDA) oder der Europäischen Arzneimittel-Agentur (EMA) ein. Diese Behörden bewerten die Daten, um sicherzustellen, dass das Medikament Vorteile bietet, die seine bekannten und potenziellen Risiken für die vorgesehene Bevölkerung überwiegen.
- Kontextanalyse: Gutachter analysieren die Zielerkrankung oder -krankheit, für die das Medikament bestimmt ist. Sie berücksichtigen die bestehende Behandlungslandschaft und wägen die Risiken des Medikaments gegen seinen Nutzen ab. Zum Beispiel kann ein Medikament zur Behandlung einer lebensbedrohlichen Krankheit ohne alternative Therapie zugelassen werden, selbst wenn die Risiken für eine nicht lebensbedrohliche Erkrankung inakzeptabel wären.
- Bewertung klinischer Daten: FDA-Prüfer bewerten die vom Arzneimittelhersteller eingereichten Informationen zu klinischem Nutzen und Risiko. Sie berücksichtigen alle Unsicherheiten, die sich aus unvollständigen oder ungenauen Daten ergeben. Typischerweise erwartet die Behörde Ergebnisse aus gut konzipierten klinischen Studien, um die Wirksamkeit und Sicherheit des Medikaments zu validieren.
- Erstellung des Artwork:
- Sobald der Etiketteninhalt genehmigt ist, besteht der nächste Schritt in der Erstellung des Etiketten-Artworks. Dies beinhaltet das Design der visuellen Elemente, des Layouts, der Schriftarten und Grafiken. Das Artwork muss den regulatorischen Richtlinien entsprechen und die Informationen auf dem Etikett genau wiedergeben.
- Das Etiketten-Artwork wird intern im Pharmaunternehmen überprüft, um Konsistenz und Compliance sicherzustellen. Es enthält Details wie Dosierungsanweisungen, Warnhinweise, Lagerbedingungen und Kontaktinformationen.
- Das finale Artwork wird den Aufsichtsbehörden zur Genehmigung vorgelegt. Dieser Schritt stellt sicher, dass die visuelle Darstellung des Etiketts den Qualitätsstandards entspricht und wichtige Informationen effektiv an die Nutzer übermittelt.
- Herstellung und Umsetzung:
- Nach der Genehmigung wird das Etiketten-Artwork zu einem integralen Bestandteil der Arzneimittelverpackung. Hersteller stellen sicher, dass die Etiketten korrekt an jeder Produkteinheit (z. B. Flaschen, Blisterpackungen, Vials) angebracht sind.
- Qualitätskontrollverfahren überprüfen, ob Etiketten den Spezifikationen entsprechen, einschließlich korrektem Inhalt, Lesbarkeit und Einhaltung der Designrichtlinien.
- Das Etikett dient als Brücke zwischen dem Pharmaunternehmen, den Regulierungsbehörden, den Gesundheitsdienstleistern und den Patienten. Es spielt eine entscheidende Rolle bei der Gewährleistung einer sicheren und wirksamen Medikamentenanwendung während des gesamten Produktlebenszyklus.
Was sind häufige Herausforderungen in der regulatorischen Kennzeichnung?
Häufige Herausforderungen bei der regulatorischen Kennzeichnung sind die Einhaltung sich entwickelnder regulatorischer Anforderungen, die Verwaltung mehrsprachiger Kennzeichnungen und die Sicherstellung der Konsistenz über verschiedene Produktportfolios hinweg. Die dynamische Natur der regulatorischen Standards für die pharmazeutische Kennzeichnung erfordert ständige Wachsamkeit, um mit den neuesten Anforderungen Schritt zu halten. Die Anpassung an sich ändernde Vorschriften und die prompte Umsetzung notwendiger Aktualisierungen von Kennzeichnungsinhalten und -formaten ist unerlässlich, um die Compliance aufrechtzuerhalten und die Patientensicherheit zu gewährleisten.
| Herausforderung | Beschreibung |
|---|---|
| Sich entwickelnde regulatorische Anforderungen | Umgang mit sich ständig ändernden Vorschriften und Richtlinien, die ständige Aktualisierungen der Kennzeichnungsdokumentation erfordern. |
| Globale Harmonisierung | Sicherstellung der Konsistenz von Produktinformationen in verschiedenen Regionen, in Übereinstimmung mit den unterschiedlichen Anforderungen verschiedener Gesundheitsbehörden. |
| Integration von Post-Marketing-Daten | Verwaltung der Einbeziehung von Post-Marketing-Sicherheits- und Wirksamkeitsdaten in die Kennzeichnung, unter Beibehaltung von Genauigkeit und Relevanz. |
| Einhaltung lokaler Kennzeichnungsstandards | Einhaltung spezifischer Kennzeichnungsstandards einzelner Länder, unter Berücksichtigung sprachlicher, kultureller und Formatierungsunterschiede. |
| Effizientes Management von Kennzeichnungsänderungen | Optimierung der Prozesse für die zeitnahe und genaue Verfolgung, Umsetzung und Dokumentation von Kennzeichnungsänderungen. |
Mehrsprachige Kennzeichnung stellt eine erhebliche Herausforderung für Pharmaunternehmen dar, die auf globalen Märkten tätig sind. Die genaue Übersetzung von Kennzeichnungsinhalten in mehrere Sprachen unter Einhaltung regionaler sprachlicher und regulatorischer Nuancen erfordert Detailgenauigkeit und robuste Übersetzungsmanagementprozesse. Die Sicherstellung von Konsistenz und Klarheit über verschiedene Sprachversionen hinweg ist entscheidend, um wichtige Informationen effektiv an unterschiedliche Patientengruppen zu übermitteln.
Die Aufrechterhaltung der Konsistenz über verschiedene Produktportfolios hinweg stellt eine weitere häufige Herausforderung bei der regulatorischen Kennzeichnung dar. Pharmaunternehmen verwalten oft mehrere Produkte mit unterschiedlichen Kennzeichnungsanforderungen, Formulierungen und Anwendungsgebieten. Um Kohärenz und Konformität über diverse Produktlinien hinweg zu erreichen und gleichzeitig die spezifischen regulatorischen Anforderungen für jedes Produkt zu erfüllen, sind effiziente Prozesse und Systeme erforderlich, die eine Einheitlichkeit bei Inhalt, Format und Botschaft der Kennzeichnung gewährleisten.
Welche wichtigen Vorschriften gibt es für die Kennzeichnung von Arzneimitteln?
Die pharmazeutische Kennzeichnung unterliegt einem komplexen Regelwerk, das die Sicherheit, Wirksamkeit und korrekte Anwendung von Medikamenten gewährleisten soll. Einige davon sind unten aufgeführt:
US FDA (US Food and Drug Administration)
Die US FDA regelt die pharmazeutische Kennzeichnung durch eine strenge Reihe von Vorschriften, die im Code of Federal Regulations (CFR) Titel 21 dargelegt sind. Diese Vorschriften verlangen, dass Etiketten umfassende Informationen bereitstellen, einschließlich Arzneimittelindikationen, Gebrauchsanweisungen, Kontraindikationen und potenziellen Nebenwirkungen. Die FDA betont die Bedeutung einer klaren, präzisen und unmissverständlichen Sprache, um die Patientensicherheit und eine fundierte Entscheidungsfindung durch Gesundheitsdienstleister zu gewährleisten. Darüber hinaus erstrecken sich die Kennzeichnungsanforderungen der FDA auf verschiedene Aspekte wie Verpackung, Beipackzettel und elektronische Kennzeichnung, um sicherzustellen, dass alle Informationen zugänglich und über verschiedene Formate hinweg standardisiert sind. Die Einhaltung dieser Vorschriften ist für die Arzneimittelzulassung und die fortgesetzte Marktpräsenz in den US zwingend erforderlich.
EMA (Europäische Arzneimittel-Agentur)
Die EMA überwacht die pharmazeutische Kennzeichnung in der EU (European Union) durch Richtlinien und Leitfäden, die darauf abzielen, die Kennzeichnung in den Member States zu harmonisieren. Die Richtlinie 2001/83/EG der Europäischen Kommission ist zentral für diese Bemühungen und legt die Anforderungen für die Zusammenfassung der Merkmale des Arzneimittels (SmPC), Packungsbeilagen und Verpackungsetiketten fest. Die EMA stellt sicher, dass die Kennzeichnung wesentliche Informationen sowohl für medizinisches Fachpersonal als auch für Patienten enthält, um eine sichere und wirksame Medikamentenanwendung in der gesamten EU (European Union) zu fördern. Darüber hinaus muss die Kennzeichnung in den Amtssprachen der Member States verfügbar sein, in denen das Medikament vermarktet wird, was das Engagement der EMA für Zugänglichkeit und patientenzentrierte Versorgung widerspiegelt.
TGA (Therapeutic Goods Administration)
In Australien ist die TGA für die Regulierung der pharmazeutischen Kennzeichnung gemäß dem Therapeutic Goods Act 1989 verantwortlich. Die Richtlinien der TGA schreiben vor, dass Arzneimittelkennzeichnungen klare, genaue und umfassende Informationen über das Produkt enthalten müssen, einschließlich seiner Inhaltsstoffe, Anwendungsgebiete, Dosierung und potenziellen Risiken. Die Kennzeichnungsanforderungen sollen die öffentliche Gesundheit schützen, indem sie sicherstellen, dass Verbraucher und medizinisches Fachpersonal die notwendigen Informationen für eine sichere und wirksame Anwendung von Medikamenten erhalten. Die TGA legt auch großen Wert auf die Lesbarkeit der Kennzeichnungen und verlangt, dass diese in verständlichem Englisch verfasst sind und wichtige Informationen deutlich sichtbar angezeigt werden, um Fehlanwendung und Medikationsfehler zu vermeiden.
Health Canada
Health Canada reguliert die pharmazeutische Kennzeichnung durch ein Rahmenwerk, das die Sicherheit und das Wohlergehen von Patienten und medizinischem Fachpersonal in den Vordergrund stellt. Der Food and Drugs Act und die zugehörigen Vorschriften legen die Anforderungen für Arzneimittelkennzeichnungen fest, die detaillierte Informationen zur Zusammensetzung des Produkts, Anwendungsgebieten, Gegenanzeigen und möglichen Nebenwirkungen enthalten müssen. Health Canada schreibt außerdem vor, dass die Kennzeichnungen zweisprachig, in Englisch und Französisch, verfasst sein müssen, um der sprachlichen Vielfalt des Landes Rechnung zu tragen. Darüber hinaus aktualisiert Health Canada regelmäßig seine Kennzeichnungsanforderungen, um neue wissenschaftliche Erkenntnisse und sich entwickelnde Anforderungen an die öffentliche Gesundheit widerzuspiegeln und so sicherzustellen, dass die Kennzeichnung relevant und wirksam bleibt, um die sichere Anwendung von Medikamenten zu fördern.
PMDA (Pharmaceuticals and Medical Devices Agency)
Die PMDA, Japans Regulierungsbehörde, überwacht die pharmazeutische Kennzeichnung gemäß dem Pharmaceutical Affairs Law und den zugehörigen Richtlinien. Die PMDA verlangt, dass Arzneimittelkennzeichnungen umfassende Informationen, einschließlich Anwendungsgebieten, Dosierungsanweisungen und möglichen unerwünschten Wirkungen, in einem Format bereitstellen, das sowohl von medizinischem Fachpersonal als auch von Patienten leicht verstanden wird. Die PMDA schreibt außerdem vor, dass die Kennzeichnungen Warnhinweise und Vorsichtsmaßnahmen enthalten, die speziell auf die japanische Bevölkerung zugeschnitten sind, unter Berücksichtigung von Faktoren wie genetischen Unterschieden und kulturellen Praktiken. Dieser Ansatz stellt sicher, dass Medikamente in Japan sicher und effektiv angewendet werden, mit einer Kennzeichnung, die auf die spezifischen Bedürfnisse des lokalen Marktes zugeschnitten ist.
NMPA (National Medical Products Administration)
In China reguliert die NMPA die pharmazeutische Kennzeichnung durch ein regulatorisches Rahmenwerk, das Genauigkeit, Klarheit und Sicherheit betont. Das Drug Administration Law of the People's Republic of China legt die Anforderungen für Arzneimittelkennzeichnungen fest, die Informationen zu Anwendungsgebieten, Dosierung, Gegenanzeigen und möglichen Nebenwirkungen des Arzneimittels enthalten müssen. Die NMPA verlangt außerdem, dass die Kennzeichnung in vereinfachtem Chinesisch dargestellt wird, um die Zugänglichkeit für die lokale Bevölkerung zu gewährleisten. Zusätzlich schreibt die NMPA vor, dass Kennzeichnungen während des Arzneimittelzulassungsprozesses einer strengen Prüfung unterzogen werden, um die Einhaltung nationaler Standards zu gewährleisten und die öffentliche Gesundheit durch die Verhinderung von Medikationsfehlern und Missbrauch zu schützen.
Wie kann ein Partner für regulatorische Angelegenheiten dabei helfen, die Kennzeichnungsvorschriften einzuhalten?
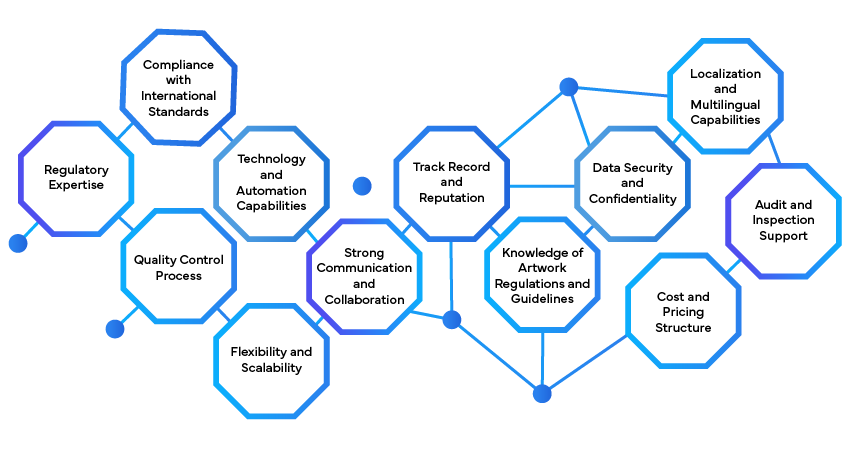
Ein Regulatorik-Partner ist maßgeblich daran beteiligt, die Einhaltung der Kennzeichnungsanforderungen zu gewährleisten, indem er spezialisiertes Fachwissen und umfassende Unterstützung anbietet. Er führt Unternehmen durch die komplexe regulatorische Landschaft und stellt sicher, dass Kennzeichnungsmaterialien – einschließlich Verpackungen, Beipackzettel und elektronische Etiketten – den spezifischen Anforderungen verschiedener Gesundheitsbehörden wie der US FDA, EMA, TGA, Health Canada, PMDA und NMPA entsprechen. Dies beinhaltet das Verständnis und die Anwendung der neuesten Vorschriften, die regional erheblich variieren können, um sicherzustellen, dass alle Produktinformationen präzise, vollständig und konform sind.
Darüber hinaus hilft ein regulatorischer Partner, den Kennzeichnungsprozess zu optimieren, indem er wichtige Dienstleistungen wie Inhaltserstellung, -prüfung und -validierung anbietet. Sie unterstützen bei der Erstellung und Überarbeitung von Kennzeichnungsinhalten, um diese an regulatorische Standards anzupassen und sicherzustellen, dass alle notwendigen Informationen enthalten sind – von Inhaltsstofflisten und Gebrauchsanweisungen bis hin zu Sicherheitshinweisen und Lagerbedingungen. Dies reduziert das Risiko von Fehlern und Auslassungen, die zu regulatorischen Verzögerungen oder Marktrücknahmen führen könnten, und beschleunigt die Markteinführungszeit für neue Produkte.

Zusätzlich unterstützt ein regulatorischer Partner Unternehmen dabei, die kontinuierliche Einhaltung der Vorschriften zu gewährleisten, indem er regulatorische Aktualisierungen überwacht und bei Bedarf Änderungen umsetzt. Sie bieten strategische Beratung zur Anpassung von Kennzeichnungen an neue Richtlinien oder aufkommende Marktanforderungen, wodurch Unternehmen Probleme bei der Nichteinhaltung vermeiden und sicherstellen, dass ihre Produkte den aktuellen Vorschriften entsprechen. Durch die Nutzung ihrer Expertise und das Bleiben auf dem Laufenden über regulatorische Änderungen hilft ein regulatorischer Partner Unternehmen, sich effizient und effektiv in der dynamischen Kennzeichnungslandschaft zurechtzufinden.
Wie können Unternehmen mit Dienstleistungen zur regulatorischen Kennzeichnung beginnen?
Um mit Dienstleistungen für die regulatorische Kennzeichnung zu beginnen, sollten Unternehmen zunächst ihre spezifischen Kennzeichnungsbedürfnisse basierend auf den Zielmärkten und regulatorischen Anforderungen bewerten. Als Nächstes sollten sie mit einem zuverlässigen Anbieter von Regulierungsdienstleistungen zusammenarbeiten, der über Fachwissen in globalen Kennzeichnungsstandards verfügt. Dieser Anbieter kann beim Entwurf, der Überprüfung und der Aktualisierung von Etiketten unterstützen, um die Einhaltung zu gewährleisten. Zusätzlich hilft die Implementierung eines zentralisierten Kennzeichnungsmanagementsystems, den Prozess zu optimieren und die Konsistenz über alle Produktetiketten hinweg sicherzustellen. Regelmäßige Audits und Aktualisierungen sind unerlässlich, um die Etiketten an sich entwickelnde Vorschriften anzupassen.
Können Dienstleistungen zur regulatorischen Kennzeichnung bei der Marktüberwachung unterstützen?
Ja, Dienstleistungen im Bereich der regulatorischen Kennzeichnung können tatsächlich bei der Marktüberwachung von Arzneimitteln nach dem Inverkehrbringen unterstützen. Diese Dienstleistungen spielen eine entscheidende Rolle bei der Unterstützung der Marktüberwachung, indem sie die Verwaltung von Kennzeichnungsaktualisierungen erleichtern, sicherheitsrelevante Kennzeichnungsänderungen bearbeiten und die Einhaltung regulatorischer Anforderungen nach der Zulassung gewährleisten. Durch die Pflege präziser und aktueller Kennzeichnungsinformationen helfen Dienstleistungen im Bereich der regulatorischen Kennzeichnung Pharmaunternehmen, auf Sicherheitsbedenken zu reagieren und notwendige Änderungen zeitnah umzusetzen, um die fortlaufende Einhaltung regulatorischer Vorschriften und die Patientensicherheit zu unterstützen.
Darüber hinaus können Dienstleistungen im Bereich der regulatorischen Kennzeichnung bei der effizienten Verbreitung aktualisierter Sicherheitsinformationen an medizinisches Fachpersonal und Patienten helfen. Im Falle neuer Sicherheitserkenntnisse oder Änderungen der Risikoprofile von Arzneimitteln können Experten für regulatorische Kennzeichnung dabei helfen, Kennzeichnungsinhalte schnell zu aktualisieren, um die neuesten Sicherheitsdaten und regulatorischen Anforderungen widerzuspiegeln. Dieser proaktive Ansatz bei der Marktüberwachung nach dem Inverkehrbringen und bei Kennzeichnungsaktualisierungen unterstützt die zeitnahe Kommunikation wichtiger Sicherheitsinformationen an Gesundheitsdienstleister und Patienten und trägt so zu einer verbesserten Pharmakovigilanz und Patientenversorgung bei.
Insgesamt bieten Dienstleistungen im Bereich der regulatorischen Kennzeichnung wertvolle Unterstützung bei der Marktüberwachung nach dem Inverkehrbringen, indem sie sicherstellen, dass Arzneimittel während ihres gesamten Lebenszyklus eine präzise und konforme Kennzeichnung aufweisen. Durch die Nutzung des Fachwissens von Regulierungsexperten und effizienter Prozesse zur Kennzeichnungsverwaltung können Unternehmen Sicherheitsaspekte nach dem Inverkehrbringen und regulatorische Verpflichtungen effektiv angehen und tragen so zur fortlaufenden Sicherheit und Wirksamkeit ihrer Produkte auf dem Markt bei.
Warum Freyr wählen?
![about-us]()
Ein Jahrzehnt Exzellenz in der regulatorischen Kennzeichnung
![about-us]()
Über 180 globale Experten für Kennzeichnungsdienstleistungen
![about-us]()
Spezialisiert auf die Erstellung und Verwaltung wesentlicher Dokumente
![about-us]()
Expertise in Prüfarztbroschüren, Kerndatenblättern und Unternehmens-Kerndatenblättern
![about-us]()
Engagiert für globale Einhaltung von Vorschriften und Präzision
![about-us]()
Nutzt KI (Künstliche Intelligenz) für eine effiziente regulatorische Navigation






Werden Sie jetzt unser Partner
Fakten auf einen Blick
Unsere Dienstleistungen








Häufig gestellte Fragen
Core Data Sheets (CDS) bieten eine konsolidierte Zusammenfassung kritischer Arzneimittelinformationen, einschließlich Indikationen, Dosierungen und Sicherheitsprofilen. Sie gewährleisten eine konsistente Kommunikation wesentlicher Details über globale Märkte hinweg, was die Einhaltung regulatorischer Vorschriften und eine fundierte Entscheidungsfindung erleichtert. CDS dienen auch als Referenz für die Erstellung lokaler Produktkennzeichnungen.
Investigational Brochures (IB) enthalten detaillierte Daten aus klinischen Studien und Informationen zur Arzneimittelentwicklung für den Prüfzweck, während Company Core Data Sheets (CCDS) wichtige Sicherheits- und Wirksamkeitsdaten für globale regulatorische Zwecke zusammenfassen und den Inhalt und die Aktualisierungen von Etiketten leiten. CCDS werden verwendet, um produktspezifische Etiketten für die Marktzulassung zu erstellen.
Künstliche Intelligenz verbessert die regulatorische Kennzeichnung, indem sie die Datenanalyse automatisiert, die Genauigkeit bei der Inhaltserstellung erhöht und die Dokumentenprüfung beschleunigt. KI-Tools optimieren Kennzeichnungsprozesse und gewährleisten die Konsistenz über verschiedene regulatorische Anforderungen hinweg. Sie helfen auch dabei, potenzielle Compliance-Probleme vorherzusagen und zu beheben.
Mehrsprachige Kennzeichnung stellt sicher, dass pharmazeutische Produkte für verschiedene Patientengruppen zugänglich sind, regionale regulatorische Anforderungen erfüllen und die Sicherheit erhöhen, indem klare, verständliche Anweisungen und Warnhinweise in mehreren Sprachen bereitgestellt werden. Dies reduziert das Risiko von Missverständnissen und Fehlern bei der Arzneimittelverabreichung.
Ein zentralisiertes Kennzeichnungsmanagementsystem koordiniert die Erstellung, Überprüfung und Aktualisierung von Kennzeichnungsdokumenten und gewährleistet so Konsistenz und Compliance über globale Märkte hinweg. Es optimiert Prozesse und pflegt genaue, aktuelle Produktinformationen. Dieses System unterstützt auch die effiziente Bearbeitung von Kennzeichnungsänderungen und regulatorischen Aktualisierungen.
Structured Product Labeling (SPL) ist ein XML-basiertes Format, das für die Arzneimittelkennzeichnung verwendet wird und Produktinformationen standardisiert und organisiert. Es gewährleistet Konsistenz und erleichtert den Datenaustausch zwischen Regulierungsbehörden und Herstellern. SPL unterstützt die effiziente Verwaltung von Kennzeichnungsinformationen über den gesamten Produktlebenszyklus hinweg.
Die Global Location Number (GLN) ist eine eindeutige Kennung, die zur Identifizierung von Standorten und Unternehmen innerhalb der Lieferkette verwendet wird. Sie hilft bei der genauen Verfolgung und Verwaltung pharmazeutischer Produkte auf globalen Märkten. GLNs gewährleisten eine präzise und effiziente Produktverteilung und Bestandsverwaltung.
Der National Drug Code (NDC) ist eine eindeutige Kennung für Arzneimittel, die von der FDA zugewiesen wird. Er hilft bei der präzisen Identifizierung von Arzneimittelprodukten und erleichtert die Bestandsverwaltung und Nachverfolgung. Der NDC ist entscheidend für eine genaue Arzneimittelabgabe und die regulatorische Berichterstattung.
Eine Investigator's Brochure (IB) enthält detaillierte Informationen zu den klinischen und präklinischen Daten eines Prüfpräparats. Sie dient dazu, Prüfärzte über die Sicherheit, Wirksamkeit und Dosierung des Arzneimittels für Studienzwecke zu informieren. Die IB unterstützt auch die ethische und fundierte Entscheidungsfindung in der klinischen Forschung.