
2 min ler
Após a descoberta inicial de impurezas de nitrosamina em medicamentos e Ingredientes Farmacêuticos Ativos (APIs) pela USFDA meados de 2018, os órgãos reguladores da UE também se juntaram a muitos outros países numa tentativa de prevenir os riscos envolvidos. Eles recolheram vários medicamentos que representam perigos à saúde devido à substância. Conhecida por suas propriedades cancerígenas, a nitrosamina pode ser prejudicial quando ingerida acima dos níveis aceitáveis para seres humanos. A substância N-nitrosodimetilamina (NDMA) tem um limite aceitável de 96 ng/dia e qualquer valor acima disso é inaceitável em medicamentos e APIs.
As nitrosaminas são formadas quando aminas secundárias, terciárias ou quaternárias reagem com um agente nitrosante. Todos os medicamentos que têm ingredientes ativos sintetizados quimicamente estão a ser verificados quanto à presença de impurezas. O Comité dos Medicamentos Uso Humano (CHMP) analisou e elaborou um relatório de avaliação. Solicitou Autorização de Introdução no Mercado (MAHs) que sigam as orientações mais recentes para a revisão de todos os medicamentos químicos e biológicos atualmente disponíveis no mercado para consumo humano.
Os MAHs são responsáveis por garantir que o processo de fabrico de todos os produtos biológicos e químicos seja revisto periodicamente para identificar qualquer contaminação. Uma vez identificada, devem ser tomadas as medidas relevantes para atenuar os riscos que ela representa. Embora as chances de tal contaminação sejam menores durante o fabrico de medicamentos químicos e biológicos, a Agência Europeia de Medicamentos (EMA) não está a correr riscos. As empresas farmacêuticas precisam garantir que os protocolos de fabrico relevantes estejam em vigor, de acordo com as EMA mais recentes EMA . Elas também são responsáveis por verificar os níveis de nitrosamina nos medicamentos disponíveis no mercado e mantê-los dentro do limite aceitável.
Após finalizar uma revisão nos termos do artigo 5.º, n.º 3, do Regulamento (CE) n.º 726/2004 (pedido de revisão) no ano passado, a EMA novas orientações para evitar a presença de contaminação por nitrosamina em medicamentos para uso humano. O processo é o mesmo para produtos autorizados a nível nacional e autorizados a nível central. A EMA, juntamente com a Direção Europeia para a Qualidade dos Medicamentos e Cuidados de Saúde (EDQM), irá implementar o artigo 5.º, n.º 3, do CHMP. Aqui está o processo que deve ser seguido para manter a conformidade com as modificações.
A orientação em três etapas para os titulares de AIM
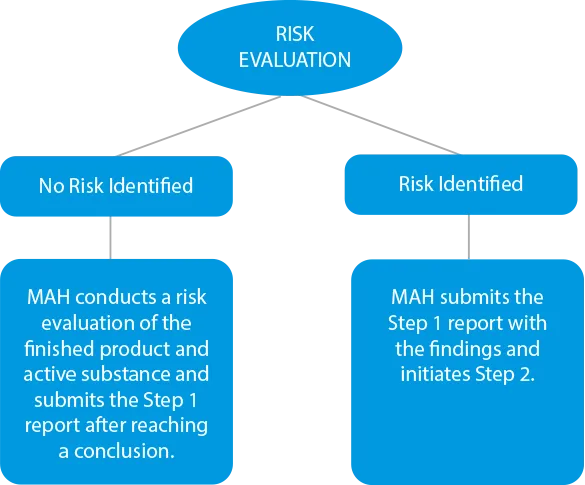
- Avaliação dos riscos - Os fabricantes têm de realizar um processo de avaliação dos riscos para identificar as substâncias activas e o produto acabado para verificar os níveis de nitrosaminas. Se houver algum caso de contaminação cruzada, o mesmo deve ser incluído no relatório de resultados. Os prazos para apresentação nesta fase foram fixados em 31 de março de 2021, para os medicamentos químicos, e em 1 de julho de 2021, para os medicamentos biológicos.
![]()
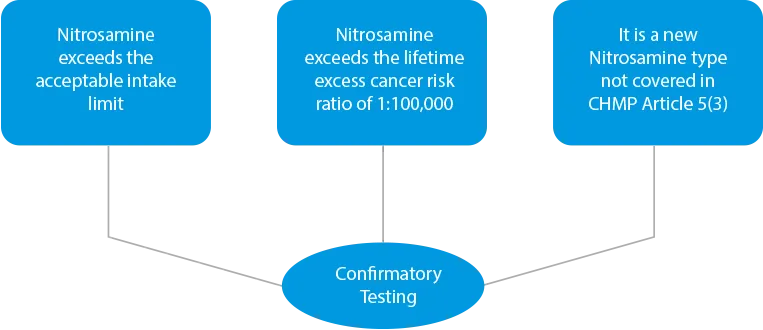
- Testes de confirmação - No caso de se constatar qualquer contaminação cruzada ou se os produtos forem identificados como de risco devido à presença de níveis mais elevados de nitrosamina, devem ser efectuados testes de confirmação. Os testes de confirmação são obrigatórios em três (03) casos.
![]()
- Autorização de Introdução no Mercado – Quando a presença de nitrosamina é detetada, dois (02) testes confirmatórios precisam ser realizados para relatar as leituras corretas à EMA. Com base nisso, os MAHs devem solicitar a alteração do processo de fabricação. Isso é feito usando os procedimentos regulatórios padrão com a ajuda de uma variação no Autorização de Introdução no Mercado . Os prazos para isso são 26 de setembro de 2022, para produtos químicos, e 1º de julho de 2023, para medicamentos biológicos.
Caminho ideal para os titulares de AIM lidarem com a conformidade com o novo nível de nitrosaminas
Uma vez que esta é a mais recente medida EMA pela EMA para mitigar os riscos associados aos níveis de nitrosamina nos medicamentos e a situações de contaminação cruzada, todo o processo pode ser bastante complexo para os titulares de autorizações de introdução no mercado (MAHs) e os fabricantes de API. Seja enviando o modelo de resposta quando a contaminação é detetada na Etapa 1 ou realizando testes consequentes, todas as fases devem estar em conformidade com as novas regras. Os MAHs precisam colaborar com especialistas regulatórios que estejam atualizados com todas as mudanças mais recentes e garantir a conformidade com as novas orientações. Escolha o parceiro certo, como Freyr evitar atrasos e erros.









