
4 min ler
Os produtos combinados, que integram medicamentos, dispositivos e/ou produtos biológicos, revolucionaram os cuidados de saúde ao oferecerem soluções terapêuticas inovadoras. No entanto, a sua natureza única apresenta desafios regulamentares significativos. Este guia abrangente explora o intrincado cenário da regulamentação de produtos combinados e fornece estratégias para navegar com sucesso por esses caminhos complexos.
Compreender os produtos combinados e o seu quadro regulamentar
Os produtos combinados são produtos médicos que combinam dois (2) ou mais componentes regulamentados — medicamentos, dispositivos ou produtos biológicos — numa única entidade. Estes produtos inovadores podem assumir várias formas, tais como stents farmacológicos, seringas pré-cheias ou adesivos transdérmicos. O quadro regulamentar para produtos combinados é regido principalmente pela Food and Drug Administration (FDA) dos Estados Unidos, com abordagens semelhantes adotadas por órgãos reguladores em todo o mundo.
O Gabinete de Produtos Combinados (OCP) FDA desempenha um papel crucial na determinação do modo de ação primário (PMOA) de um produto combinado, que determina o caminho regulatório e o FDA principal FDA responsável pela revisão. O Centro de Avaliação e Investigação de Medicamentos (CDER), o Centro de Dispositivos e Saúde Radiológica (CDRH) e o Centro de Avaliação e Investigação Biológica (CBER) colaboram no processo de revisão, enfatizando a necessidade de uma abordagem integrada para garantir a segurança e a eficácia dos produtos combinados.
Determinação da via de regulação
Uma (1) das etapas mais críticas no desenvolvimento de uma estratégia regulamentar para produtos combinados é a determinação da via regulamentar adequada. Esta decisão baseia-se principalmente no PMOA do produto. Eis algumas considerações fundamentais:
- Modo de ação primário (PMOA): Identificar o modo de ação único que proporciona a ação terapêutica mais importante do medicamento combinado.
- Atribuição do centro principal: Com base no PMOA, o produto será atribuído ao CDER, CDRH ou CBER para análise primária.
- Tipo de submissão regulatória: Dependendo do centro líder, a submissão pode assumir a forma de uma submissão de novo medicamento submissão NDA), submissão de licença biológica submissão BLA) ou aprovação pré-comercialização (PMA).
- Pedido de Designação (RFD): Se a PMOA não for clara, os patrocinadores podem apresentar um RFD ao OCP para uma determinação formal.
- Pré-Solicitação de Designação (Pré-RFD): O OCP pode receber um Pré-RFD para obter feedback informal e não vinculativo sobre questões de classificação e jurisdição.
A compreensão destes factores é crucial para o desenvolvimento de uma estratégia regulamentar eficaz, adaptada à combinação específica de produtos.
Navegar nos processos de revisão pré-comercialização
O processo de revisão pré-comercialização de produtos combinados pode ser complexo, exigindo um planeamento e execução cuidadosos. Eis as principais estratégias para navegar eficazmente neste processo:
- Envolvimento precoce com os reguladores: Inicie discussões com a FDA do processo de desenvolvimento por meio de reuniões pré-apresentação. Essas interações podem fornecer orientações valiosas sobre requisitos regulatórios, desenhos de estudos e possíveis desafios.
- Plano de Desenvolvimento Abrangente: Desenvolver um plano robusto que aborde os aspectos únicos do seu produto combinado, incluindo a forma como as partes constituintes interagem e os seus efeitos combinados na segurança e eficácia.
- Abordagem de testes integrados: Conceber estudos pré-clínicos e clínicos que avaliem os componentes individuais e o produto combinado completo. Esta abordagem deve avaliar potenciais interações e efeitos cumulativos.
- Considerações sobre o sistema de qualidade: Implemente um sistema de qualidade que esteja em conformidade com os regulamentos relativos a medicamentos (21 CFR 210/211) e dispositivos (21 CFR 820), conforme apropriado para o seu produto.
- Engenharia de Factores Humanos: Incorporar estudos de factores humanos para avaliar as interações dos utilizadores com o produto combinado, garantindo uma utilização segura e eficaz.
- Gestão de riscos: Desenvolver um Plano de Gestão de Risco abrangente Plano de Gestão de Risco aborde os riscos potenciais associados a cada componente e à sua combinação.
Ao adotar estas estratégias, os promotores podem simplificar o processo de análise antes da comercialização e aumentar a probabilidade de sucesso regulamentar.
Enfrentar os desafios pós-comercialização
As responsabilidades regulatórias para produtos combinados vão além da aprovação de comercialização. A vigilância pós-comercialização eficaz e a conformidade são cruciais para o sucesso a longo prazo. Considere as seguintes estratégias:
- Farmacovigilância integrada: Implementar um sistema de farmacovigilância robusto que registe os eventos adversos relacionados com os componentes do medicamento e do dispositivo do produto combinado.
- Estudos pós-comercialização: Planear e realizar estudos pós-comercialização para recolher dados adicionais de segurança e eficácia, especialmente para novos produtos combinados.
- Manutenção do sistema de qualidade: Atualizar e manter continuamente o seu sistema de qualidade para garantir a conformidade com os regulamentos relevantes para os componentes de medicamentos e dispositivos.
- Gestão de alterações: Estabelecer um processo simples para gerir as alterações pós-aprovação, tendo em conta o potencial impacto nos aspectos do medicamento e do dispositivo do produto.
- Informação regulamentar: Mantenha-se informado sobre a evolução dos regulamentos e documentos de orientação relacionados com produtos combinados para garantir a conformidade contínua.
Ao abordar proactivamente estes desafios pós-comercialização, os fabricantes podem manter a conformidade regulamentar e a segurança dos produtos ao longo do seu ciclo de vida.
Considerações sobre a regulamentação global
À medida que o mercado de produtos combinados se expande globalmente, a compreensão dos requisitos regulamentares internacionais torna-se cada vez mais importante. Considere estas estratégias para o sucesso regulamentar global:
- Harmonização regulamentar: Aproveitar os esforços de harmonização internacional, tais como os do Fórum Internacional de Reguladores de Dispositivos Médicos (IMDRF), para simplificar as submissões regulamentares globais.
- Requisitos específicos do mercado: Pesquisar e compreender os requisitos específicos para produtos combinados nos mercados-alvo, uma vez que os regulamentos variam significativamente entre países.
- Apresentações simultâneas: Considerar apresentações simultâneas a várias agências reguladoras para acelerar o acesso ao mercado global, quando apropriado.
- Ensaios clínicos globais: Conceber ensaios clínicos que cumpram os requisitos de várias agências reguladoras para apoiar aplicações de marketing globais.
- Parcerias internacionais: Colaborar com especialistas em regulamentação locais ou parceiros em mercados-alvo para navegar por regulamentos específicos do país e considerações culturais.
Ao adotar uma perspetiva global no desenvolvimento de estratégias regulatórias, os fabricantes podem expandir reach seu reach de mercado reach produtos combinados de forma mais eficaz.
Conclusão
Navegar pelas vias regulamentares para produtos combinados requer uma compreensão abrangente de quadros regulamentares complexos e planeamento estratégico.
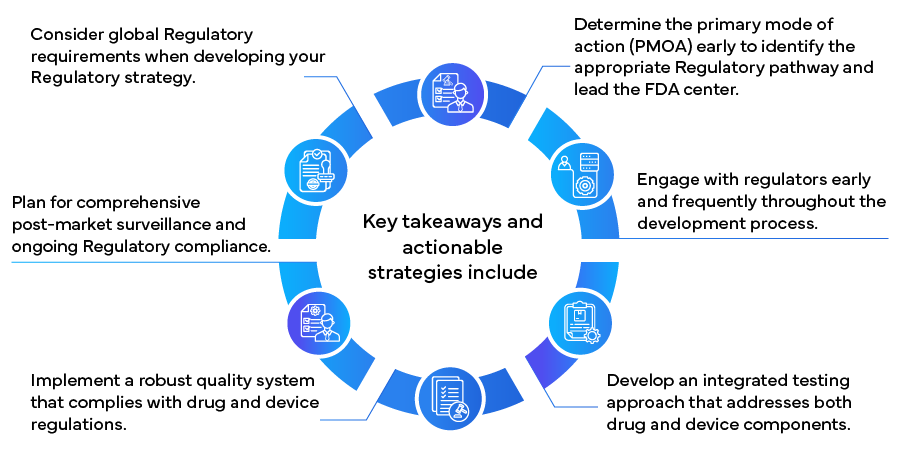
Ao implementar estas estratégias, os fabricantes podem navegar mais eficazmente no complexo panorama regulamentar dos produtos combinados, acelerando potencialmente o tempo de colocação no mercado e assegurando a conformidade e o sucesso a longo prazo. À medida que o campo dos produtos combinados continua a evoluir, manter-se informado sobre as alterações regulamentares e manter a flexibilidade nas abordagens regulamentares será crucial para o sucesso contínuo neste sector de cuidados de saúde inovador e em rápido crescimento.









