
3 min ler
O protocolo de validação é definido como um plano documentado para testar um dispositivo médico para confirmar que o processo de produção utilizado para fabricar o produto cumpre os requisitos específicos do utilizador, técnicos e regulamentares. Isto inclui uma revisão das variáveis do processo e das limitações operacionais e a análise dos resultados dos testes em condições de utilização reais.
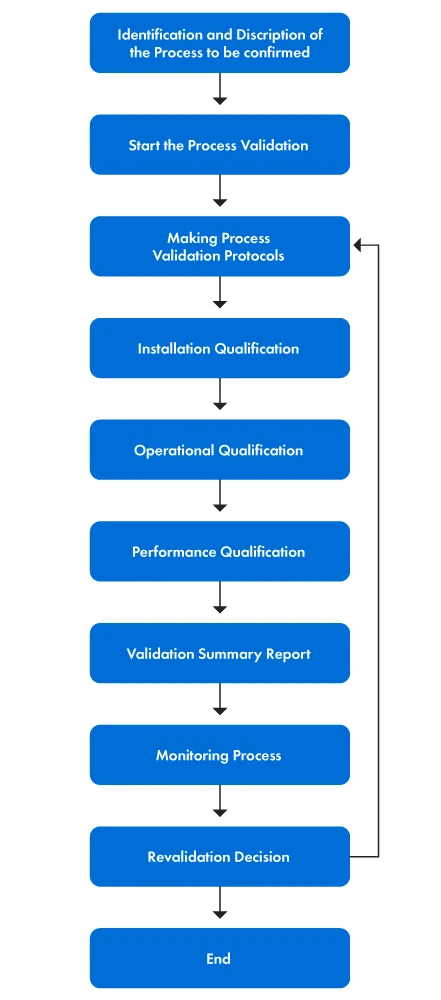
O processo de validação envolve várias acções concretas. As etapas são elucidadas da seguinte forma:
- Primeiro, a equipa de validação é formada e a cada membro são atribuídas funções e responsabilidades específicas. A finalidade da validação do processo é fornecer uma declaração clara dos objectivos de validação e definir o âmbito das actividades de validação, especificando os aspectos do dispositivo médico que estão a ser validados. A equipa compreende então os princípios subjacentes ao processo para identificar parâmetros específicos e resultados desejados.
- Em segundo lugar, são estabelecidos os critérios de avaliação e aceitação, juntamente com a seleção dos métodos de teste, ferramentas e técnicas de análise estatística adequados. De seguida, são elaborados os protocolos de validação do processo e implementadas a Qualificação de Instalação (QI), a Qualificação Operacional (QO) e a Qualificação de Desempenho (QD).
- Por último, são determinados controlos contínuos do processo e medidas de monitorização para garantir a validação contínua do processo. Sempre que necessário, a revalidação é efectuada para manter a precisão e a eficácia do processo de validação.
A figura 1 abaixo apresenta uma representação passo a passo do processo de validação.
Figura 1: As fases do processo de validação
PVP
Devido à vasta gama de volumes de produção e complexidades de fabrico, existem inúmeras abordagens para realizar a validação de processos. No entanto, os regulamentos da United States Food and Drug AdministrationUSFDA) e da ISO 13485 fornecem sugestões limitadas sobre métodos específicos. No entanto, uma fonte amplamente reconhecida e autorizada para a validação de processos de dispositivos médicos é um documento de orientação do Global Harmonization Task Force (GHTF), atualmente denominado International Medical Device Regulators Forum (IMDRF), publicado em 2004. Continua a ser a principal referência, mesmo no sítio Web oficial da USFDA.
De acordo com o documento de orientação, é formada uma equipa de validação para criar um Plano de Validação do Processo (PVP) detalhado. Os protocolos de validação de processos incluem um esquema detalhado sobre como implementar IQ, OQ, PQ e revalidação. O PVP deve conter os seguintes elementos:
- Definir o dispositivo e determinar a abordagem de validação.
- Identificar os elementos que necessitam de validação.
- Realização de actividades no local designado.
- Definição do âmbito da documentação.
- Criar um calendário para as actividades de validação.
- Desenvolver um calendário geral.
- Manter uma lista exaustiva e referências às validações internas e externas que foram efectuadas.
O protocolo de validação é escrito antes da realização das actividades de validação. Deve ser preparado pela equipa de validação e aprovado pelo departamento competente. O objetivo de um protocolo de validação é definir os roteiros de teste que devem ser seguidos para garantir que os processos e equipamentos estão prontos para fabricar produtos de dispositivos médicos seguros e eficazes.
Um relatório analítico que contém informações juntamente com as análises, explicações e recomendações necessárias faz parte do protocolo de validação. Estes registos são ainda analisados para garantir que os dois (02) critérios seguintes são cumpridos:
- Cumprimento das normas regulamentares.
- Todos os registos e dados gerados são analisados quanto aos resultados, à adequação e à exaustividade.
A figura 2 abaixo representa o PVP e os vários processos nele envolvidos.
Figura 2: O PVP e os seus requisitos
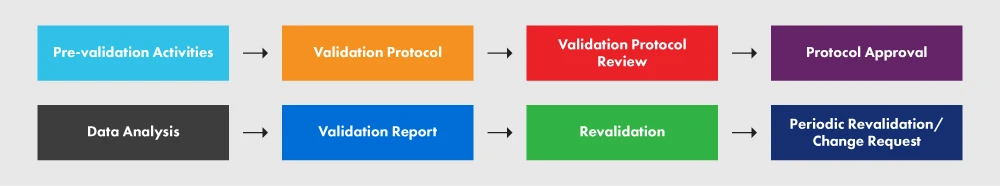
Um protocolo adequadamente redigido fornece diretrizes, políticas e procedimentos claros a serem cumpridos durante a validação do processo. Ele engloba aspectos como instalações, equipamentos, métodos e treinamento. O protocolo especifica as entradas e os limites do processo, bem como os passos essenciais para a execução bem sucedida do projeto de validação do processo. Embora o esquema que se segue não englobe todos os elementos necessários no seu protocolo, dá-lhe uma visão geral do nível de detalhe necessário. Recomendamos vivamente que siga o documento de orientação para uma melhor compreensão do processo.
- Título da página
- Produtos a abranger
- Equipamento/Processo a validar
- Geral
- Objectivos
- Documentos de referência
- Plano de validação
- QI
- OQ
- PQ
- Equipamento de medição/ensaio e calibração
- Manutenção de equipamentos
- Revalidação
- Página de aprovação/assinatura da equipa de validação
A gestão de operações desempenha um papel crucial na manutenção de um desempenho ótimo, monitorizando as medidas-chave, revendo os métodos e procedimentos de trabalho e tomando medidas imediatas quando surgem quaisquer problemas. Nos casos em que existam problemas, poderá ser necessário revalidar um processo parcial ou mesmo totalmente. De acordo com a Secção 820.75(c) do Regulamento do Sistema de Qualidade USFDA (QSR), a revalidação do processo deve ser considerada nestas circunstâncias: "Quando ocorrem alterações ou desvios do processo, o fabricante deve rever, avaliar e efetuar a revalidação conforme apropriado. Estas actividades devem ser documentadas."
Os possíveis factores que desencadeiam a revalidação do processo incluem alterações às especificações, métodos, procedimentos, software, desenhos, componentes-chave, escalonamento de lotes, alterações de localização, alterações de equipamento e semelhantes. Além disso, a implementação de Acções Corretivas e Preventivas (CAPA) também pode servir como um fator de revalidação do processo. As principais razões para a revalidação são as seguintes:
- Alterações efectuadas no processo.
- Tendência negativa na qualidade, deterioração súbita da qualidade ou um pico de reclamações dos clientes.
- Grande expansão da capacidade da linha.
- Alterações na conceção.
- Alterações na embalagem dos produtos.
- Transferência de um processo para outra instalação.
- Alterações no submissão .
Para saber mais sobre os protocolos de validação e a sua importância no domínio do fabrico de dispositivos médicos, us Mantenha-se informado! Mantenha-se em conformidade!









