
4 Minuten lesen
Mit der Umsetzung der endgültigen Regelung für die Qualitätsmanagementsystemvorschriften (QMSR) durch die US und Arzneimittelbehörde (USFDA) im Jahr 2024 müssen Hersteller von Medizinprodukten die Änderungen berücksichtigen, um ihre Produkte auf dem US-amerikanischen Markt vertreiben zu können.
Mit dieser Vorschrift werden die Qualitätssystemvorschriften (QSR) der USFDAaktualisiert, indem sie an die ISO 13485:2016 angepasst werden, die internationale Norm für QMS für Medizinprodukte. Die Hersteller von Medizinprodukten haben eine zweijährige Übergangsfrist für die Anpassung, was es für die Organisationen notwendig macht, die neuen Anforderungen anzupassen, um eine Nichteinhaltung zum Zeitpunkt der Inspektion zu vermeiden.
Was ist QMSR?
Das FDA ist ein optimierter Ansatz für QMS-Anforderungen, der eine Aktualisierung der früheren QSR-Struktur darstellt. Diese Angleichung ist von entscheidender Bedeutung, da sie die globale Compliance für Hersteller vereinfacht, insbesondere für diejenigen, die weltweit tätig sind. Durch diese Harmonisierung können Unternehmen die regulatorischen Anforderungen sowohl in den US in anderen Märkten auf wesentlich einheitlichere Weise erfüllen.
Der QMSR schreibt Verbesserungen in den Bereichen Risikomanagement, Produktdesign und Überwachung nach dem Inverkehrbringen vor. Diese Rolle kann zu mehr Komplexität führen, aber sie bietet den Herstellern auch die Möglichkeit, ihre Qualitätsverfahren zu standardisieren, was wiederum die Sicherheit der Produkte erhöht und zu einer besseren Dokumentation führt, die bei USFDA entscheidend sein kann.
Die wichtigsten Änderungen des QMSR
- Harmonisierung mit ISO 13485: Dies ist der wichtigste Schritt, der es Herstellern von Medizinprodukten ermöglicht, international anerkannte Normen zu akzeptieren. Die USFDA hat erkannt, dass viele Unternehmen, die Medizinprodukte herstellen, bereits mit der ISO 13485 übereinstimmen, wodurch sich doppelter Aufwand vermeiden lässt.
- Der Schwerpunkt des Risikomanagements liegt auf dem Risikomanagement während des gesamten Lebenszyklus eines Medizinprodukts. Hersteller von Medizinprodukten müssen ein wirksames Risikomanagement, eine wirksame Risikokontrolle und ein wirksames Risikomanagement aufweisen.
- Produktauslegung und -kontrollen: Im Rahmen des QMSR wurden die Auslegungskontrollen ausgeweitet, um sicherzustellen, dass die Hersteller von Medizinprodukten die Bedürfnisse der Anwender, die Sicherheit des Produkts und die Leistungskriterien, die bei den USFDA im Mittelpunkt stehen, vollständig berücksichtigen.
- Post Market Surveillance: Die Unternehmen müssen das System zur Überwachung nach dem Inverkehrbringen verbessern. Dies wird die Hersteller dazu veranlassen, Informationen über die Sicherheit und Wirksamkeit von Produkten zu sammeln, die dazu beitragen, Probleme schnell zu erkennen und schnell zu identifizieren.
- Dokumentation und Aufbewahrung von Unterlagen: Dies ist die letzte Vorschrift, die den Schwerpunkt auf die Dokumentation legt. Gründliche / ordnungsgemäße Aufzeichnungen sind bei der Inspektion von entscheidender Bedeutung.
Schritte zur Vorbereitung auf die Inspektion durch die US und Arzneimittelbehörde (FDA):
Da die Inspektion bis 2026 dauert, haben die Unternehmen zwei Jahre Zeit, um ihre Qualitätssysteme an das QMSR anzupassen. Allerdings kann es riskant sein, bis zur letzten Minute zu warten.
Schritte für USFDA im Rahmen des QMSR für Branchen, deren derzeitiges QMS auf dem QSR basiert:
- Durchführen einer Lückenanalyse: Dies ist der erste Schritt, wenn das derzeitige Qualitätssystem von den neuen QMSR-Anforderungen abweicht. Eine gründliche Lückenanalyse wird dazu beitragen, die Bereiche zu ermitteln, die aktualisiert werden müssen, wie z. B. Risikomanagement, Überwachung nach dem Inverkehrbringen und Entwurfskontrollen.
- Aktualisierung des Risikomanagementverfahrens: Stellen Sie sicher, dass die Risikomanagementaktivitäten mit dem gesamten Lebenszyklus Ihres Produkts vom Entwurf bis zur Überwachung nach dem Inverkehrbringen kombiniert werden.
- Überprüfen Sie die Designkontrolle: Die Hersteller sollten sicherstellen, dass der Entwurfsprozess solide und gut dokumentiert ist. Validieren Sie, dass der Entwurfsprozess robust und gut dokumentiert und vollständig in Ihr Qualitätsmanagementsystem integriert ist.
- Verstärkung der Überwachung nach dem Inverkehrbringen: Einführung von Systemen zur Überwachung der Leistung des Produkts nach seiner Markteinführung. Dies könnte die Einrichtung von Mechanismen für das Kundenfeedback, die Sammlung von klinischen Daten und deren genaue Verfolgung umfassen.
- Schulung und Dokumentation: Schulung des Personals zu den neuen Anforderungen, insbesondere derjenigen, die mit dem Qualitätsmanagement und der Einhaltung der Vorschriften befasst sind. Dadurch wird sichergestellt, dass alle Dokumentationsprozesse mit den Erwartungen des QMSR übereinstimmen.
- Zertifizierung durch Dritte: Wenn Ihr Unternehmen noch nicht nach ISO 13485 zertifiziert ist, sollten Sie dies jetzt in Erwägung ziehen. Eine Zertifizierung nach ISO 13485 kann Ihnen einen Vorsprung bei der Erfüllung der Anforderungen der USFDA verschaffen und Ihre Glaubwürdigkeit auf den globalen Märkten erhöhen.
Navigieren in der zweijährigen Übergangszeit
Um die Übergangszeit effektiv zu nutzen und die Einhaltung der USFDA über Qualitätsmanagementsysteme (QMSR) zu gewährleisten, sollten die Hersteller einen proaktiven Ansatz wählen. Hier ist ein vorgeschlagener Fahrplan:
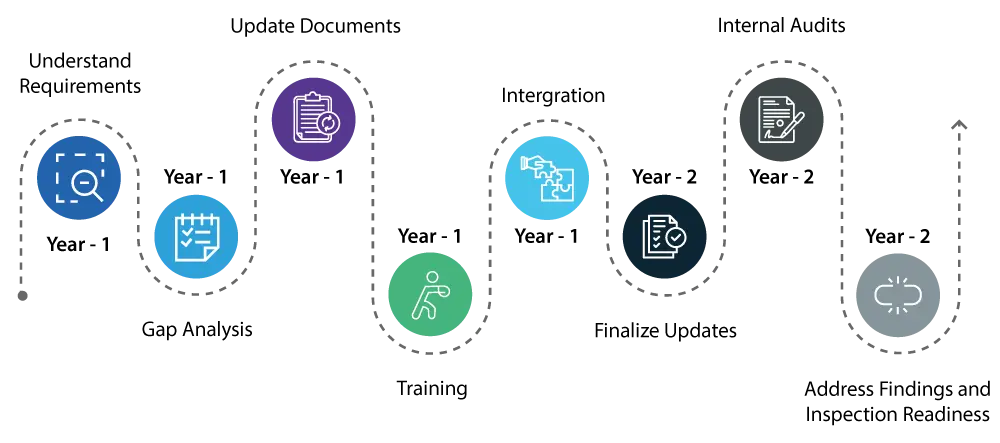
Jahr 1:
- Verstehen Sie die Anforderungen: Beginnen Sie mit einem gründlichen Verständnis der durch die QMSR eingeführten Änderungen. Dazu gehört eine detaillierte Überprüfung der neuen Anforderungen und ihrer Unterschiede zur bestehenden Verordnung über Qualitätssicherungssysteme (QS).
- Die Lücken kennen: Führen Sie eine umfassende Lückenanalyse durch, um die Bereiche innerhalb Ihres derzeitigen Qualitätssystems zu ermitteln, die aktualisiert werden müssen, um den neuen QMSR-Normen zu entsprechen.
- Aktualisierung der Dokumente / Korrekturmaßnahmen: Initiieren Sie die notwendigen Aktualisierungen Ihres Qualitätssystems und konzentrieren Sie sich dabei auf Bereiche wie Risikomanagement und Entwurfskontrollen, die wichtige Bestandteile des QMSR sind.
- Schulung: Beginnen Sie mit der Schulung Ihrer Mitarbeiter zu den neuen Vorschriften, um sicherzustellen, dass alle Beteiligten über die Änderungen informiert sind und ihre Rolle bei der Einhaltung der Vorschriften verstehen.
- Integration: Beginnen Sie damit, die neuen QMSR-Anforderungen in Ihre täglichen Abläufe zu integrieren, um den Übergang reibungsloser zu gestalten.
Jahr 2:
- Fahren Sie mit der Umsetzung von Änderungen an Ihrem Qualitätssystem fort und stellen Sie sicher, dass alle Aktualisierungen vollständig integriert und betriebsbereit sind.
- Führen Sie gründliche interne Audits durch, um zu überprüfen, ob die Aktualisierungen wirksam sind und ob Ihr Qualitätssystem vollständig mit den QMSR-Anforderungen übereinstimmt.
- Kümmern Sie sich umgehend um alle Feststellungen aus den internen Audits, um sicherzustellen, dass alle Aspekte Ihres Qualitätssystems den Vorschriften entsprechen.
- Am Ende des zweiten Jahres sollte Ihr Qualitätssicherungssystem vollständig mit dem QMSR übereinstimmen, und Sie sollten auf die USFDA vorbereitet sein und darauf vertrauen können, dass es keine größeren Probleme geben wird.
Indem sie diesen Fahrplan befolgen, können Hersteller nicht nur die Anforderungen der USFDA erfüllen, sondern auch ein robustes, effizientes, standardisiertes und weltweit anerkanntes Qualitätssystem aufbauen. Dieser proaktive Ansatz wird dazu beitragen, einen reibungslosen Übergang zu den neuen Vorschriften zu gewährleisten und die höchsten Qualitäts- und Sicherheitsstandards für Medizinprodukte aufrechtzuerhalten.
Schlussfolgerung: Proaktive Schritte unternehmen
Bei der Vorbereitung auf USFDA im Rahmen der neuen QMSR-Regelung geht es nicht nur um die Vermeidung von Strafen, sondern auch um die Verbesserung der Sicherheit und Wirksamkeit von Produkten. Durch die Harmonisierung mit ISO 13485 setzt die USFDA höhere Erwartungen, bietet aber auch einen Weg zu einer strafferen globalen Compliance. Hersteller, die frühzeitig mit der Anpassung beginnen, sollten sich auf Schlüsselbereiche wie Risikomanagement und Überwachung nach dem Inverkehrbringen konzentrieren und sicherstellen, dass ihre Qualitätssysteme auf dem neuesten Stand und robust sind, damit sie nicht nur die Erwartungen der Regulierungsbehörden erfüllen, sondern auch ihren Wettbewerbsvorteil auf dem Markt ausbauen können.
