
5 min. de leitura
O software para dispositivos médicos na Coreia do Sul é utilizado para diagnosticar, tratar e monitorizar pacientes no sistema de saúde moderno. Inclui software incorporado que é integrado em dispositivos médicos e software autónomo que pode ser utilizado em PCs, dispositivos móveis e serviços baseados na Web. O Ministério da Segurança Alimentar e dos Medicamentos (MFDS) da Coreia do Sul é responsável pela regulamentação do software para dispositivos médicos e por garantir a sua segurança e eficácia. A 05 de julho de 2023, o MFDS estabeleceu critérios para a aprovação e inspeção de software para dispositivos médicos; estes regulamentos fornecem uma estrutura que os peticionários civis podem seguir quando submetem software para aprovação ou revisão.
Os regulamentos abordam uma variedade de assuntos, incluindo o escopo da submissão, requisitos de documentação técnica e relatórios de verificação de conformidade. Existem normas e diretrizes internacionais que se aplicam ao software de dispositivos médicos, além das diretrizes da MFDS, como a norma 62304 da Comissão Eletrotécnica Internacional (IEC) para processos do ciclo de vida do software e a orientação da Administração de Alimentos e Medicamentos dos Estados Unidos (US FDA) sobre aplicações médicas móveis.
Plano de desenvolvimento de software e análise de requisitos
- O Plano de Desenvolvimento de Software descreve a abordagem global ao desenvolvimento de software, incluindo especificações, métodos e ferramentas de desenvolvimento. Também abrange a verificação, a gestão dos riscos dos dispositivos médicos, a gestão da configuração e a documentação.
- A análise de requisitos estabelece os requisitos de software para dispositivos médicos, incluindo medidas de controlo de riscos e métodos de verificação. Ao planear e analisar cuidadosamente o processo de desenvolvimento de software, os programadores podem garantir que o software resultante cumpre as normas necessárias de segurança e eficácia.
- O Relatório de Verificação da Conformidade do Software inclui um esboço do plano de desenvolvimento do software, o número de controlo do documento do fabricante e uma visão geral da análise dos requisitos. Ao aderir a estas diretrizes, o software para dispositivos médicos pode ser desenvolvido com confiança, sabendo que foi submetido a testes rigorosos e que cumpre as normas necessárias de segurança e eficácia.
Verificação e validação de software para dispositivos médicos
- A Verificação de Software para Dispositivos Médicos garante que o software cumpre os requisitos especificados.
- A validação de software para dispositivos médicos garante que o software satisfaz as necessidades do utilizador e a(s) utilização(ões) prevista(s).
- O Relatório de Verificação e Validação de Software para Dispositivos Médicos descreve o processo de verificação e validação, incluindo o nome do produto, a revisão e os nomes das pessoas who examinaram e aprovaram o relatório. O relatório pode variar, dependendo das caraterísticas do software, mas deve incluir uma descrição do software, os métodos de verificação e validação utilizados e os resultados dos testes.
Ambiente Operativo e Software de Proveniência Desconhecida (SOUP)
- Se o software depender de hardware específico, como é o caso do software incorporado, o documento técnico deve descrever as especificações do hardware.
- No entanto, se o software for autónomo e desenvolvido para funcionar em hardware de uso geral, o ambiente operacional deve ser descrito na matéria-prima. Tal inclui as especificações mínimas recomendadas, como o Microsoft Windows 10 ou superior.
- Além disso, se o software para dispositivos médicos incluir Software comercial de proveniência desconhecida (SOUP), deve ser criado um ambiente operacional para garantir o funcionamento correto. Descrevendo cuidadosamente o ambiente de funcionamento e abordando qualquer SOUP, os programadores podem garantir que o seu software para dispositivos médicos é seguro e eficaz para a utilização pretendida.
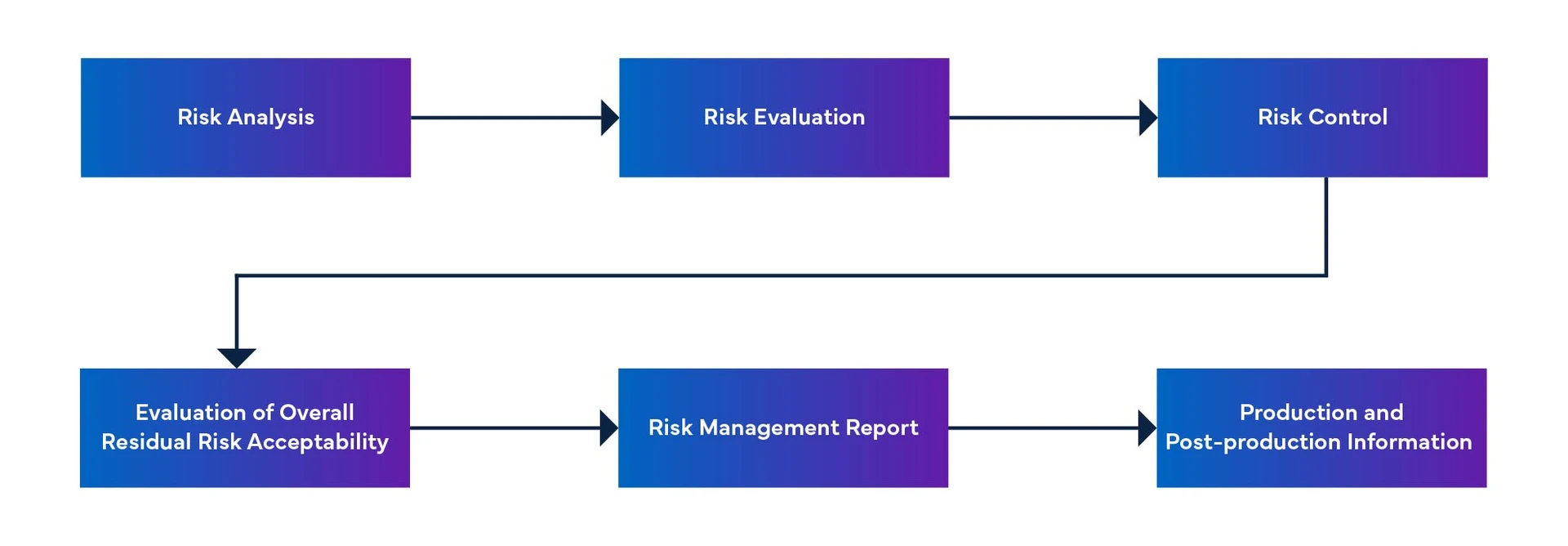
Gestão do risco dos dispositivos médicos e requisitos de documentação
- O processo de gestão do risco do software as a medical device inclui a identificação de situações perigosas, o estabelecimento de medidas de controlo do risco, a verificação dessas medidas e a gestão das alterações ao software.
- O documento MFDS-RM de gestão do risco de software fornece informações sobre a gestão do risco de software.
- Além disso, os requisitos de documentação são essenciais para garantir que o software cumpre as normas de segurança e eficácia necessárias.
- O plano de desenvolvimento do software, a análise dos requisitos de software dos dispositivos médicos e os relatórios de verificação e validação do software devem ser incluídos na documentação.
- O Relatório de Verificação da Conformidade do Software descreve os requisitos de documentação; inclui também uma descrição dos documentos aplicáveis e o número de controlo dos documentos do fabricante.
Figura 1: Processo de Gestão do Risco dos Dispositivos Médicos
Anomalias não resolvidas e acções corretivas para o software SaMD
- O documento MFDS-PR (Resolução de Problemas de Software) descreve o processo de resolução de problemas de software, que inclui a comunicação, análise, implementação e verificação de problemas.
- O documento também inclui uma lista de problemas não resolvidos, bugs, defeitos e anomalias, bem como uma avaliação de risco residual para o sistema de software.
- As acções corretivas tomadas para resolver estes problemas devem ser documentadas no plano de manutenção do software, que é estabelecido de acordo com o processo de manutenção do software.
- O documento MFDS-maintenance fornece informações sobre SaMD e a resolução de problemas.
Requisitos de revisão e apresentação de documentos técnicos para o software SaMD
Os principais documentos de análise durante o processo de análise são os dados de desempenho, o relatório de confirmação da conformidade e os dados de verificação e validação do software para dispositivos médicos, a Especificação de Conceção do Software (SDS), a Declaração de Requisitos do Software para dispositivos médicos (SRS) e os relatórios de verificação e validação. O Relatório de Confirmação da Conformidade e o Relatório de Verificação e Validação do Software para Dispositivos Médicos devem ser apresentados.
Gestão de riscos de software para dispositivos médicos
- Identificar os riscos potenciais associados ao software e à sua utilização.
- Avaliar a gravidade dos riscos associados a estes perigos.
- Implementação de medidas de controlo dos riscos para minimizar a probabilidade de danos.
- Acompanhamento e revisão da eficácia destas medidas de controlo dos riscos.
- Documentar todas as actividades e decisões de gestão do risco dos dispositivos médicos.
Num sistema de software, os itens de software são divididos em partes mais pequenas, incluindo itens de software detalhados. Quando um item não pode ser mais dividido, é designado por unidade. O sistema permite a decomposição ao nível da unidade, ajudando a determinar o nível de segurança para cada item de software. Ao juntar estes itens de software, é possível determinar o nível de segurança para todo o sistema de software.
Figura 2: Desmontagem e integração de software de dispositivos médicos
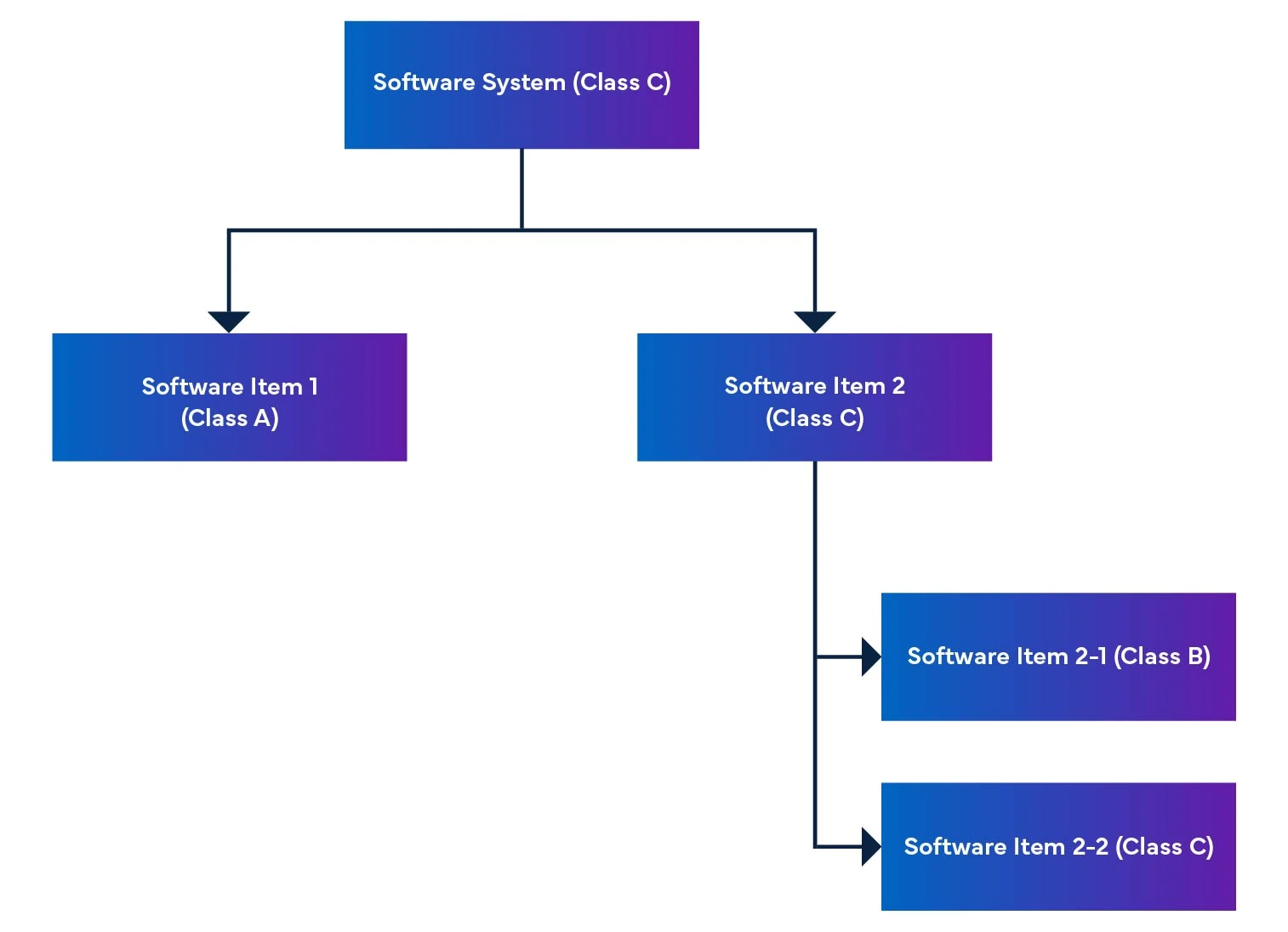
O regulamento também menciona a classificação de segurança do software, que é uma classificação para identificar os riscos do software SaMD (ver Quadro 1).
Quadro 1: Definição de classificação de segurança
| Classificação | Definição de classe de segurança de software de dispositivo médico |
| Classe A | Não há possibilidade de ferimentos ou lesões corporais. |
| Classe B | É provável que ocorram ferimentos menos graves (ferimentos ligeiros). |
| Classe C | Possibilidade de ferimentos graves ou morte. |
Gestão da configuração de software
- Manutenção de documentação exacta e actualizada para todas as versões, alterações e actualizações de software.
- Assegurar que toda a documentação é devidamente analisada e aprovada.
- Implementação de procedimentos para gerir as alterações de configuração do software.
- Documentar todas as actividades e decisões de gestão da configuração do software.
Manutenção de software
- Testar e monitorizar regularmente o software para garantir que continua a ser seguro e eficaz para a utilização a que se destina.
- Implementação de procedimentos para resolver quaisquer problemas que possam surgir, incluindo correcções de erros e actualizações de software.
- Documentar todas as actividades e decisões de manutenção de software.
Resolução de problemas
- Identificar a causa principal do problema.
- Aplicação de medidas corretivas para resolver o problema.
- Documentar todo o processo de resolução de problemas para referência futura.
Seguindo as diretrizes acima, os programadores podem garantir que quaisquer problemas com o seu software para dispositivos médicos são devidamente tratados e documentados, e que o software cumpre os requisitos necessários para aprovação ou exame.
Se é um fabricante de dispositivos médicos que pretende estar em conformidade com as normas de software para dispositivos médicos da Coreia do Sul, os especialistas em Regulamentação da Freyrpodem guiá-lo através do intrincado panorama regulamentar do país. Asseguramos que os seus dispositivos estão em conformidade com os mais recentes regulamentos da Coreia do Sul para uma conformidade perfeita. us para saber mais!









