
2 min ler
FDA US FDA um documento de orientação que ajudará a indústria e os funcionários da Agência de Saúde (HA) a determinar quando uma alteração de software num dispositivo médico exige que o fabricante apresente e obtenha a FDA para uma nova notificação pré-comercialização (510(k)). Esta orientação visa melhorar a previsibilidade, consistência e transparência do processo de tomada de decisão sobre «quando apresentar», fornecendo uma abordagem menos onerosa e descrevendo o quadro regulamentar, as políticas e as práticas subjacentes a tal decisão, especificamente relacionadas com alterações de software. Vamos descobrir a FDA em detalhe.
Princípios FDA e fluxograma FDA
Com o objetivo de auxiliar os fabricantes de dispositivos médicos na aplicação dos princípios fundamentais, o documento fornece um fluxograma, esclarecimentos adicionais e exemplos necessários para a tomada de decisões relativas a uma nova notificação pré-comercialização 510(k) para uma alteração de software feita num dispositivo já aprovado nos US. Além disso, deve-se seguir vários princípios orientadores ao utilizar este guia para determinar se deve ser apresentada uma nova 510(k) para alterar um dispositivo existente. Alguns deles são amplamente conhecidos e derivados da atual política FDA (k) FDA , enquanto outros são necessários para utilizar o esquema lógico mencionado nesta orientação. De acordo com a orientação, o esquema fornecido não pode cobrir todas as possíveis complexidades relacionadas a tais alterações e como elas afetam a decisão. Portanto, para determinar a necessidade de uma nova notificação pré-comercialização 510(k), os fabricantes de dispositivos médicos devem considerar os princípios gerais e o fluxograma resumidos abaixo.
- Alterações que afectam a segurança ou a eficácia de um dispositivo
- Avaliação inicial baseada no risco
- Consequências indesejadas das alterações
- Utilização da gestão do risco
- Papel dos ensaios (actividades de verificação e validação) na avaliação da possibilidade de uma alteração afetar significativamente a segurança e a eficácia
- Avaliação de alterações simultâneas para determinar se é necessária a apresentação de um novo 510(k)
- Dispositivo comparativo apropriado e o efeito cumulativo das alterações
- Exigência de documentos (21 CFR Parte 820)
- Apresentações 510(k) para dispositivos modificados
- Determinações de equivalência substancial
- A apresentação de um novo 510(k) é provavelmente necessária se um fabricante modificar o seu dispositivo para afetar a segurança ou a eficácia do mesmo. No entanto, as alterações que não se destinam a afetar a segurança ou a eficácia de um dispositivo devem ser avaliadas.
- Para determinar se uma alteração ou modificação pode afetar significativamente a segurança ou a eficácia, o fabricante deve começar por realizar uma avaliação baseada no risco para saber se a alteração pode afetar positiva ou negativamente a segurança ou a eficácia do dispositivo. Esta avaliação baseada no risco deve identificar e analisar todos os novos riscos e alterações nos riscos existentes resultantes da alteração do dispositivo e conduzir a uma decisão inicial sobre a necessidade de apresentação de um novo 510(k).
- Por vezes, existem consequências adicionais não intencionais ou não planeadas que podem ser desencadeadas durante a apresentação de software. O fluxograma deve avaliar essas consequências para determinar se é necessária a apresentação de um novo 510(k).
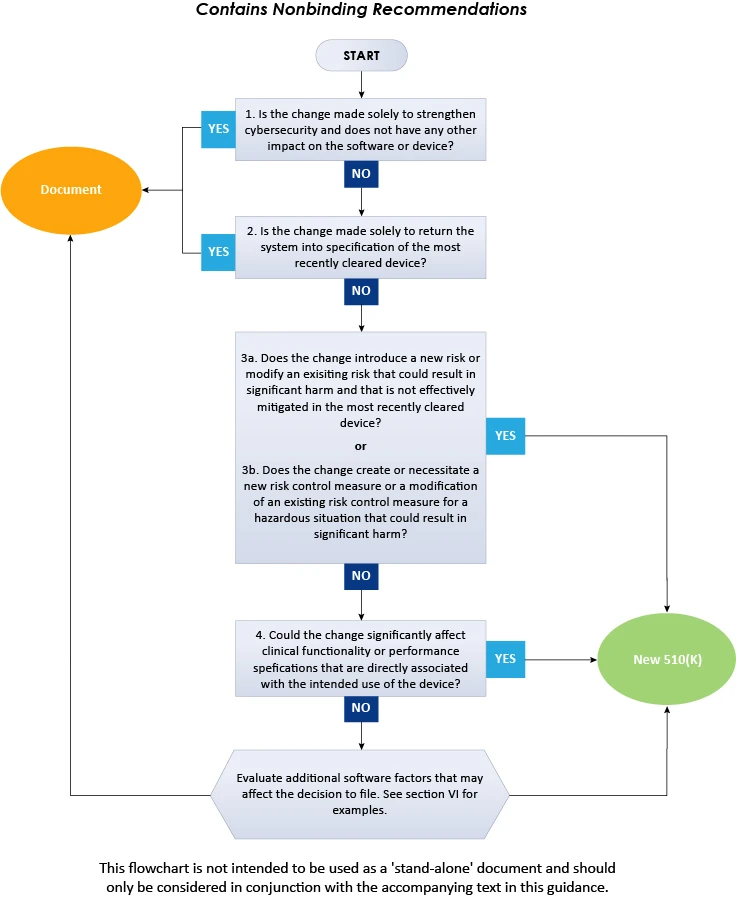
O fluxograma acima ilustra um procedimento passo a passo a ser seguido para decidir sobre a submissão 510(k) para alterações de software em dispositivos existentes. Em conclusão, a presente FDA descreve em detalhes a abordagem a ser seguida pelos fabricantes de dispositivos médicos ao decidir se as alterações de software em um dispositivo médico existente exigem a submissão de um novo 510(k). Para obter mais informações sobre a FDA , consulte Freyr, uma especialista comprovada em regulamentação. Mantenha-se informado. Mantenha-se em conformidade.









