5 min. de leitura
Submissões previstas para 24 de setembro de 2016.
Agora que a segunda fase da conformidade com a UDI, para dispositivos médicos Classe III I/LS/LS, foi implementada, muitos fabricantes de dispositivos, especialmente do tipo Classe II, estão a questionar-se sobre a melhor forma de se prepararem para o prazo de envio de dados de dispositivos Classe II, em 24 de setembro de 2016. Para os alertar, na Freyr, identificámos alguns dos pré-requisitos que devem considerar para alinhar os dispositivos Classe II com a conformidade com a exigência da UDI FDA.
A nova regulamentação exige que todos os dispositivos médicos de classe II sejam rotulados e embalados com um identificador único de dispositivo (UDI) e inseridos na Base de Dados Global de Identificação Única de Dispositivos (GUDID) FDA. Dada a volatilidade dos requisitos de conformidade, aliada a prazos de submissão mais curtos, o desafio para os fabricantes de dispositivos é conhecer os detalhes dos processos de conformidade. Ao mesmo tempo, eles devem garantir que nenhum dos atributos-chave do dispositivo seja esquecido ao reunir os dados dispersos do dispositivo em diferentes sistemas e reconciliá-los em planilhas para criar relatórios de conformidade.
Para ajudar os fabricantes de dispositivos a navegar facilmente por esse processo de conformidade complexo e urgente, sem erros, Freyr os seguintes pré-requisitos a serem seguidos.
Determine a data de conformidade com a UDI: Desde que FDA sua regra final, algumas das datas de conformidade dos dispositivos foram alteradas e prorrogadas. Para planear meticulosamente com antecedência as estratégias e os processos de conformidade e evitar alterações precipitadas de última hora, os rotuladores devem determinar a data exata de conformidade.
Data de conformidade dos dispositivos da classe II Requisitos de conformidade 24 de setembro de 2016Os dispositivos da classe III que devam ser rotulados com uma UDI devem ostentar uma UDI como marca permanente no próprio dispositivo se este se destinar a ser utilizado mais do que uma vez e a ser reprocessado antes de cada utilização. Os rótulos e as embalagens dos dispositivos médicos da classe II devem ostentar um UDI As datas nos rótulos destes dispositivos devem ser formatadas conforme necessário O software autónomo de classe II deve fornecer o seu UDI conforme exigido Os dados relativos aos dispositivos da classe II que têm de ser rotulados com uma UDI devem ser apresentados na base de dados GUDID Para a maioria dos dispositivos, a data de conformidade para a marcação direta é diferente dos outros requisitos. Com base na categoria do seu produto, quer se destine a ser reutilizado ou reprocessado, determine a data de conformidade da UDI de marcação direta, conforme indicado aqui:
Datas de conformidade da marcação direta Categoria de dispositivo - Reutilizado e Reprocessado 24 de setembro de 2015 Dispositivos de suporte de vida e de apoio à vida, independentemente da classe do dispositivo 24 de setembro de 2016 Dispositivos da classe III e dispositivos licenciados ao abrigo do Public Health Service Act 24 de setembro de 2018 Dispositivos da classe II 24 de setembro de 2020 Dispositivos da classe I e dispositivos não classificados Avaliar a necessidade de marcação direta do número UDI: Todos os dispositivos médicos que sejam utilizados mais do que uma vez ou que devam ser reprocessados antes de cada utilização devem ter uma marcação direta da UDI. A exceção é para os dispositivos implantáveis que não requerem marcação direta de acordo com a regra UDI. Os dispositivos de utilização única, mesmo que sejam reprocessados, também não são obrigados a ostentar um UDI permanente - 21 CFR 801.45(d)(3). Assim, avalie a necessidade de Marcação Direta com base na categoria de dispositivos médicos que fabrica.
- Plano de conformidade global: Reveja os FDA para que os seus produtos específicos estejam em conformidade. Realize uma análise completa das lacunas para descobrir deficiências relacionadas com dados ou tecnologia para lidar com alguns dos principais desafios no processo de cumprimento dos FDA rigorosos FDA . Alguns dos desafios podem ser a obtenção de informações DI ou PI e o tratamento de grandes volumes de dados não estruturados de fontes díspares, etc. Em vez de trabalhar até altas horas da madrugada para reconciliar todos os dados de dispositivos médicos no último minuto, planeie com antecedência uma conformidade abrangente por meio de sistemas e ferramentas validados que oferecem suporte à integração, qualidade e gestão de dados.
![]()
Obter o número de identificação e a filiação na agência: A UDI é composta pelo Identificador do Dispositivo (DI – número único baseado na versão ou modelo do dispositivo) e pelo Identificador do Produto (PI – inclui número do lote, número de série ou data de validade). A parte DI da UDI servirá como chave primária para pesquisar informações sobre o dispositivo no GUDID. Para atribuir o DI, a FDA três agências emissoras – GS1, HIBCC e ICCBBA. Nesse cenário, os rotuladores devem obter a adesão a uma das agências para obter o número DI, que precisa ser inserido no GUDID FDA
![]()
Identificador Atributos Agências emissoras UDI DI (Device Identifier - Static Data)
Necessário para ser sincronizado com o GUDIDNúmero único de
Fabricante
Marca do dispositivo
Modelo do dispositivo
GSI
HIBCC
ICCBBAPI (Product Identifier - Dynamic Data)
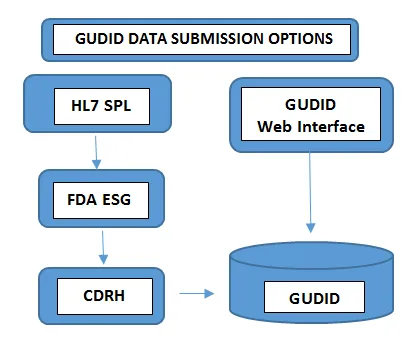
Obrigatório em todos os níveis da embalagemNúmero de lote, número de série, data de fabrico, data de expiração, - Apresentar os dados: A forma como os dados são enviados para o GUDID varia de acordo com o volume de portfólios de produtos que você lida. Fabricantes de dispositivos com um número mínimo de dispositivos optam por enviar as informações UDI manualmente através da interface web gratuita do GUDID FDA. Nesse caso, apenas um registo DI pode ser enviado por vez através de uma interface web segura do GUDID.No outro caso, os fabricantes com um número maior de portfólios de produtos optam pela opção de envio HL7 SPL para recolher dados eletronicamente e converter os dados consolidados para o formato SPL antes de enviá-los para o Electronic Submission Gateway (ESG) FDA, usando o número DUNS. Observe que a conta GUDID não é por tipo de envio. A conta serve para identificar você, o rotulador, para permitir o envio de dados do dispositivo através de ambas as opções.
![]()
- Criar uma conta GUDID: Um rotulador/fabricante de dispositivos precisa de uma ou mais contas GUDID com base no número de funções a serem atribuídas; para citar algumas, coordenador GUDID, utilizador de entrada de dados, etc. Mas para autorizar cada função para entrada de dados, o fabricante precisa de uma aprovação da FDA da criação da conta. O processo de criação de uma conta GUDID apropriada envolve o envio de um pedido por e-mail à FDA o qual o requerente, ou seja, você, receberá um documento de pedido de conta para preencher. Depois de enviar o documento preenchido para a FDA e-mail, a agência analisará o formulário e enviará um e-mail com as informações de login da conta GUDID.


A implementação da UDI é um processo complexo e demorado. Durante o curso, ao cumprir os requisitos da UDI FDA, os fabricantes de dispositivos médicos enfrentam muitos desafios relacionados à gestão, integração e envio de dados. Com o prazo de conformidade para dispositivos da Classe II a apenas um ano de distância, Freyr que as empresas comecem a trabalhar para isso agora mesmo.
Para orientar a sua organização neste complexo processo de conformidade, Freyr o melhor dos dois mundos: uma solução de software UDI sob demanda e totalmente configurável,Freyr , bem como um Centro de Excelência (CoE) que ofereceosmelhoresserviços UDIda categoria, econômicos e personalizáveis, desenvolvidos com base nos seus requisitos exclusivos e exigentes.









