2 min ler
Um «dispositivo predicado» é um dispositivo médico que foi previamente aprovado pela US and Drug Administration (FDAUS e já se encontra no mercado, servindo como ponto de referência para novos dispositivos médicos que procuram aprovação através do processo de autorização510(k) FDA.
Deve ser provado que o dispositivo em causa é, pelo menos, tão seguro e eficaz como o dispositivo de referência, em termos da sua utilização prevista e das suas caraterísticas tecnológicas. Esta comparação é conhecida como determinação de "equivalência substancial".
Um novo dispositivo não precisa de ser idêntico ao dispositivo de referência para ser substancialmente equivalente a este último.
Como identificar um dispositivo predeterminado?
A base de dados FDAfornece um código de produto de três letras para cada classificação de dispositivo. A base de dados FDA (k) contém informações sobre todos os dispositivos aprovados através doprocesso 510(k). Depois de obter o código de produto de três letras, pode obter uma lista de todos os produtos, todas as empresas e o nome comercial de todos os concorrentes ou potenciais concorrentes que deseja analisar. Pode então fazer uma análise e comparação aprofundadas para restringir um dispositivo predicado.
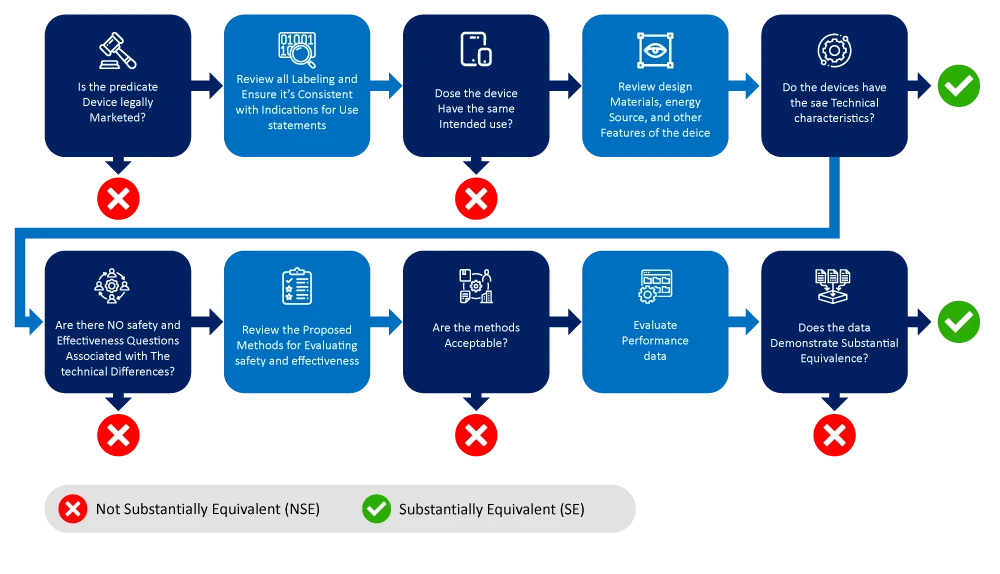
Abaixo está um fluxograma que descreve o processo de identificação e restrição de um dispositivo predicado.
Factores a considerar na determinação do(s) dispositivo(s) de referência
- Utilização prevista: A utilização prevista após o dispositivo anterior deve ser semelhante à do novo dispositivo. Por exemplo, se o novo dispositivo se destinar a ser utilizado para monitorização cardíaca, o dispositivo de referência deve também ser um dispositivo de monitorização cardíaca.
- Caraterísticas tecnológicas: O dispositivo de referência deve ser idêntico ao novo dispositivo em termos de caraterísticas tecnológicas. Veja-se, por exemplo, a conceção, os materiais utilizados e o método de funcionamento, que devem ser semelhantes.
- Biocompatibilidade: As avaliações da biocompatibilidade de um dispositivo ou componente médico não se devem limitar às matérias-primas utilizadas no dispositivo e no processo de fabrico, devendo também ser considerados outros produtos químicos. Este fator, no entanto, não se aplica aos DIV.
- Tecnologia de ponta: O dispositivo de referência não deve ser antiquado e deve representar a tecnologia médica mais recente.
O dispositivo predicado é um fator-chave para determinar se um novo dispositivo médico pode ser introduzido no mercado através da via 510(k). A escolha do dispositivo predicado errado pode resultar num processo de aprovação regulamentar mais dispendioso e demorado, enquanto a escolha do dispositivo predicado correto pode ajudar a reduzir o custo e o tempo necessários para colocar um novo dispositivo médico no mercado. Se o dispositivo predicado não for adequado, pode resultar em atrasos e despesas adicionais.
Para obter assistência com o processo de submissão 510(k) do seu dispositivo médico, agende uma chamada com os especialistas Freyr , que podem ajudá-lo a navegar pelos procedimentos. Mantenha-se informado. Mantenha-se em conformidade.









