
3 min ler
510(k) é uma submissão pré-comercialização feita à FDA demonstrar que o dispositivo a ser comercializado é tão seguro e eficaz, ou seja, substancialmente equivalente a um dispositivo comercializado legalmente (predicado). Dispositivos com risco moderado são obrigados a submeter uma notificação 510(k), que inclui uma minoria de dispositivos Classe I e III e uma maioria de dispositivos Classe II.
Existem três (03) tipos de programas 510(k), Tradicional, Abreviado e Especial. A via de segurança e desempenho foi introduzida em 2019 e construída sobre o programa abreviado. O programa eSTAR introduzido em 2020 permite uma submissão abrangente de dispositivos médicos através de um formulário PDF interativo.
Quem precisa de uma certificação 510(k)?
510(k) é essencialmente o nome do processo/caminho que os fabricantes de dispositivos médicos que pretendem comercializar os seus dispositivos de risco moderado a elevado nos US para demonstrar que o produto a ser comercializado é tão seguro e eficaz quanto um dispositivo comercializado legalmente.
Segue-se o processo passo a passo para obter uma autorização 510(k).
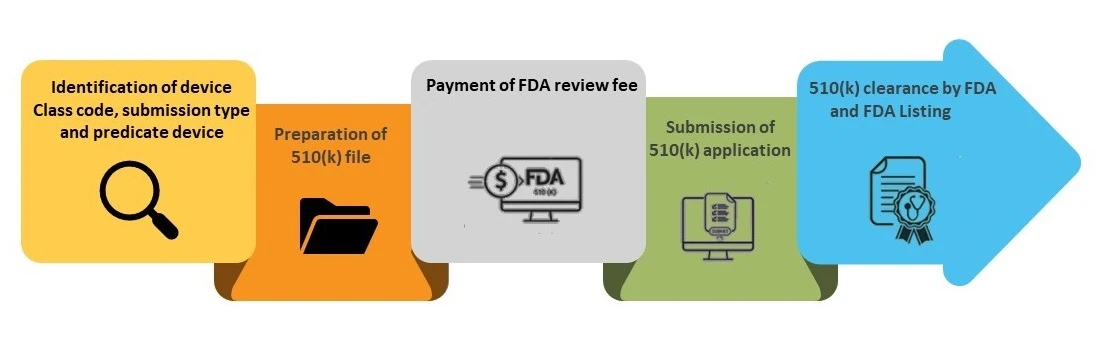
Passo 1- Identificação do código da classe do dispositivo, do tipo de apresentação e do dispositivo associado
- Identifique o código do produto e o número da regulamentação - Para determinar os requisitos do teste 510(k), é necessário primeiro identificar o código do produto e o número da regulamentação. É possível iniciar uma pesquisa na FDA para encontrar o número de regulamentação de 7 dígitos cuja identificação corresponda ao uso pretendido do dispositivo em questão.
- O código FDA é composto por três (03) letras. As informações relativas à classificação do produto, descrição da regulamentação e requisitos de BPF podem ser localizadas utilizando este código.
- Seleção do tipo de submissão - O submetente pode escolher um dos três (03) tipos de submissão mencionados anteriormente. O 510(k) tradicional é para submissões pela primeira vez, o 510(k) especial é para fabricantes de dispositivos médicos que desejam submeter alterações a um dispositivo existente e o 510(k) abreviado pode ser escolhido quando o dispositivo está em conformidade com as normas consensuais voluntárias estabelecidas. No caso de um 510(k) abreviado, o submetente deve basear-se nos documentos FDA .
- Identificação do dispositivo de referência - Um fabricante de dispositivos médicos tem de provar que o dispositivo que pretende comercializar tem a mesma utilização prevista e as mesmas caraterísticas técnicas que o dispositivo legalmente comercializado, também conhecido como dispositivo de referência. Se existirem diferenças nas caraterísticas técnicas, o transmitente deve provar que não existem preocupações de segurança e eficácia associadas a essa diferença.
Etapa 2- Preparação do ficheiro 510(k)
O próximo passo é preparar o arquivo 510(k), as orientações e as informações, que estão disponíveis no FDA . Ele inclui a lista de verificação de aceitação para todos os três (03) tipos de programas 510(k) e um microsite intitulado Conteúdo para 510(k), que inclui informações sobre declarações para indicações de uso, comparação de equivalência substancial e rotulagem proposta, entre outras informações úteis.
Etapas do processo de apresentação de 510(k)
Passo 3 - Pagamento da taxa FDA
Todos os tipos de pedidos 510(k) estão sujeitos à taxa de utilização. Para o exercício de 2023, a taxa normal para 510(k) é de $19.870. Para as empresas certificadas pelo Centro de Diagnóstico e Saúde Radiológica (CDRH), também conhecidas como pequenas empresas, a taxa é de $4.967. A taxa está sujeita a alterações no próximo ano fiscal.
Etapa 4 - Apresentação do submissão 510(k)
O transmitente pode enviar uma cópia eletrónica (eCopy) ou uma submissão eletrónica do modelo e recurso de submissão (eSTAR) antes da comercialização através do portal CDRH.
A partir de 1 de outubro de 2023, todos os pedidos de autorização 510(k), a menos que estejam isentos de acordo com as orientações finais, devem ser apresentados sob a forma de pedidos electrónicos utilizando o eSTAR.
Após o envio do 510(k), é atribuído um número de controlo único, conhecido como «número 510(k)» ou «número K». FDA duas verificações: uma para confirmar se a taxa de utilização adequada foi paga e outra para verificar se foi fornecida uma eCopy ou eSTAR válida.
- No dia 07, FDA uma carta de confirmação caso a taxa de utilização adequada tenha sido paga e uma cópia eletrónica ou eSTAR válida tenha sido fornecida. Caso contrário, a FDA uma carta de retenção para questões não resolvidas.
- No 15.º dia, FDA uma análise de aceitação. FDA o requerente se o 510(k) foi aceite para análise substantiva ou colocado em espera com recusa de aceitação (RTA).
- No 60.º dia, FDA uma revisão substantiva. FDA FDA , através de uma interação substantiva
, que FDA prosseguir com uma revisão interativa ou FDA o 510(k) será colocado em espera e que serão solicitadas informações adicionais.
Etapa 5 - FDA e listagem na base de dados FDA (k)
O objetivo da FDA anunciar a sua decisão sobre as Alterações às Taxas de Utilização de Dispositivos Médicos (MDUFA) em 90 FDA . FDA são os dias civis entre a data em que o 510(k) foi recebido e a data da decisão da MDUFA, excluindo os dias em que a submissão ficou em espera devido a um pedido de informações adicionais. As decisões da MDUFA para submissões 510(k) incluem conclusões de substancialmente equivalente (SE) ou não substancialmente equivalente (NSE).
Quando uma decisão é tomada, FDA uma carta de decisão ao requerente por e-mail. Um submissão 510(k) submissão recebe uma carta de decisão SE é considerado «aprovado». Em seguida, é listado na base de dados 510(k) juntamente com as indicações para o uso do dispositivo médico e o resumo 510(k) ou a declaração 510(k) como anexos.
Pode-se concluir que um planeamento cuidadoso e uma execução meticulosa, por meio de documentação completa e um entendimento profundo do ambiente regulatório, são cruciais para o sucesso da submissão do 510(k) à FDA.
Para obter assistência com o processo de submissão 510(k) do seu dispositivo médico, pode escrever us sales@freyrsoltions.com ou agendar uma chamada com us nossos especialistas, que podem ajudá-lo a navegar pelos procedimentos. Mantenha-se informado. Mantenha-se em conformidade.









