Übersicht über die Registrierung von Medizinprodukten beiFDA US FDA
Die Vereinigten Staaten von Amerika (USA) sind bekannt für ihren stark regulierten Markt für Medizinprodukte mit klar definierten Zulassungsverfahren und Anforderungen. Die ersten Vorschriften für Medizinprodukte in den USA stammen aus dem Jahr 1976 und wurden im Laufe der Zeit weiterentwickelt. Sie werden vom Center for Devices and Radiological Health (CDRH) der Food and Drug Administration (FDA) reguliert. Freyr hat zahlreiche Medizinproduktehersteller dabei unterstützt, die Anforderungen des ZulassungsverfahrensFDA US FDA zu erfüllen.
![]()
Regulierungsbehörde: Lebensmittel- und ArzneimittelbehördeFDA)![]()
Verordnung: Titel 21 Code of Federal Regulations (21 CFR) Teile 800 - 1299![]()
Regulierungspfad: Benachrichtigung vor dem Inverkehrbringen oder Zulassung vor dem Inverkehrbringen oder De-Novo-Klassifizierung![]()
Bevollmächtigter Vertreter: U.S. Agent![]()
QMS-Anforderung: Qualitätssystem-Verordnung (QSR) (21 CFR Teil 820)![]()
Bewertung der technischen Daten: Zentrum für Produkte und Radiologische Gesundheit![]()
Gültigkeit der Lizenz: Unbegrenzt![]()
Kennzeichnungsvorschriften: 21 CFR Teil 801![]()
Format der Einreichung: Papier & CD/DVD![]()
Sprache: Englisch
Klassifizierung von Medizinprodukten USA
Die FDA teilt Medizinprodukte in drei risikobasierte Kategorien ein: Klasse I, Klasse II und Klasse III, wobei Produkte der Klasse I als Produkte mit geringem Risiko gelten und Produkte der Klasse III mit hohem Risiko verbunden sind. Die Registrierungsanforderungen und -wege variieren je nach Produktklasse.
| Geräteklasse | Risiko | Weg der Registrierung zur Genehmigung |
|---|---|---|
| I | Geringes Risiko | 510(k) befreit |
| II | Mäßiges Risiko (mit Prädikat) | Anmeldung vor dem Inverkehrbringen/510(k) |
Mäßiges Risiko (ohne Prädikatgerät) | De-Novo-Antrag | |
| III | Hohes Risiko | Vorab-Zulassung (PMA) |
U.S. FDA
Unternehmen ohne lokale Niederlassungen in den USA müssen einen FDA benennen, der den Hersteller vertritt. Der FDA muss entweder in den USA wohnen oder einen Geschäftssitz in den USA unterhalten. Die vom Vertreter zu erfüllenden Aufgaben sind von derFDA Rahmen der CFR-Vorschriften vorab festgelegt worden.
Navigieren Siedurch die häufig gestellten Fragen (FAQs) zu US .
Interaktive Treffen mit der US FDA
FDA US FDA die Hersteller durch verschiedene Arten vonQ-Submission-Meetings, um unterschiedliche Ziele zu erreichen. Solche Treffen mit der Behörde vor Beginn oder während der Entwicklung eines Produkts und vor Einreichung der Anträge auf ZulassungFDA beiFDA US FDA helfen den Herstellern, die Zeitpläne und Kosten für die Markteinführung der Produkte zu optimieren.
Registrierung von Medizinprodukten USA
Die Produkte können von der CDRH und der FDA auf einem der verschiedenen Registrierungswege zugelassen werden. Sie sind aufgeführt als:
Medizinprodukte der Klasse I:Produkte der Klasse I sind in der Regel von der GMP- und 510(k)-Einreichungspflicht befreit und bedürfen keiner vorherigen Genehmigung durch die US FDA in den USA vermarktet zu werden. Andere Anforderungen wie die Registrierung des Herstellers, die Produktlistung, die UDI, das PMS usw. müssen vom Hersteller erfüllt werden.
Medizinprodukte der Klasse II: Produktemit mittlerem Risiko, für die 510(k)-zugelassene Referenzprodukte existieren, können sich für eine 510(k)-Vorab-Marktzulassungsanzeige (PMN) entscheiden, die auch als510(k)-Registrierung bezeichnet wird. Das betreffende Produkt muss eine wesentliche Gleichwertigkeit (Substantial Equivalence, SE) mit den identifizierten und angegebenen Referenzprodukten nachweisen. Dieser Weg ist der am weitesten verbreitete Weg für die Registrierung von Produkten in den USA. Hersteller von Produkten mit mittlerem Risiko ohne Referenzprodukte können bei der US FDA eine KlassifizierungFDA De-Novo-Anträge beantragen.
Medizinprodukte der Klasse III:Hersteller von Medizinproduktender Hochrisikoklasse III müssen bei derUS FDA einen Antrag auf Marktzulassung (Pre-Market Approval, PMA) einreichen. Die Produkte müssen einer detaillierten klinischen Bewertung unterzogen werden, und der Hersteller muss detaillierte Daten zur Sicherheit und Wirksamkeit aus klinischen Studien vorlegen. DieUS FDA im Rahmen der Bewertung eine QMS-InspektionFDA , bevor sie eineMarktzulassungfür das Produkt erteilt.
Nicht-CDRH-Registrierungen von Medizinprodukten
An einigen Grenzprodukten, die in anderen Ländern als Medizinprodukte gelten, wie z. B. chirurgische Atemschutzgeräte, Desinfektionsmittel und Kombinationsprodukte, sind aufgrund ihrer Anwendungsgebiete andere Behörden beteiligt, wie z. B. das Centre for Disease Control (CDC), das National Institute for Occupational Safety and Hazards (NIOSH), die Environmental Protection AgencyEPA, das Centre for Biological Evaluation and Research (CBER) und das Centre for Drug Evaluation and Research (CDER).
Anforderungen an die Einhaltung der Vorschriften nach der Zulassung für Medizinprodukte
Alle Hersteller von Produkten müssen die unten aufgeführten Anforderungen nach der Zulassung erfüllen:
- Voraussetzung für die Registrierung und Listung: Betriebe aller Produktklassen müssen in der FURL-Datenbank registriert werden, und das Produkt muss nach Erhalt der Zulassung und vor dem Inverkehrbringen in den USA aufgelistet werden. Einige Produkte, wie z. B. Strahlenschutzgeräte, müssen weitere Anforderungen erfüllen, wie z. B. eine Zulassungsnummer, bevor sie in die USA eingeführt werden können.
- Eindeutige Geräteidentifikation: Alle Geräteklassen müssen die Vorschriften zur eindeutigen Gerätekennzeichnung (Unique Device Identification, UDI) erfüllen, um in den USA vermarktet werden zu können.
- Einrichtungsgebühren: Der Hersteller muss die jährlichen Gebühren für die Einrichtung entrichten, um seine Registrierung aufrechtzuerhalten und weiterhin Produkte in den USA zu vermarkten. Die US FDA die Gebührenstruktur für kleinere Unternehmen mit einem gültigen Small Business Certificate reduziert.
- Qualitätsaudits: BeiGeräten, die nicht von den GMP-Vorschriften ausgenommen sind,FDA dieUS FDA jederzeit die Produktionsstätte auf Einhaltung der Qualitätssystemvorschriften (QSR) gemäß 21 CFR 820.
Prozessablauf
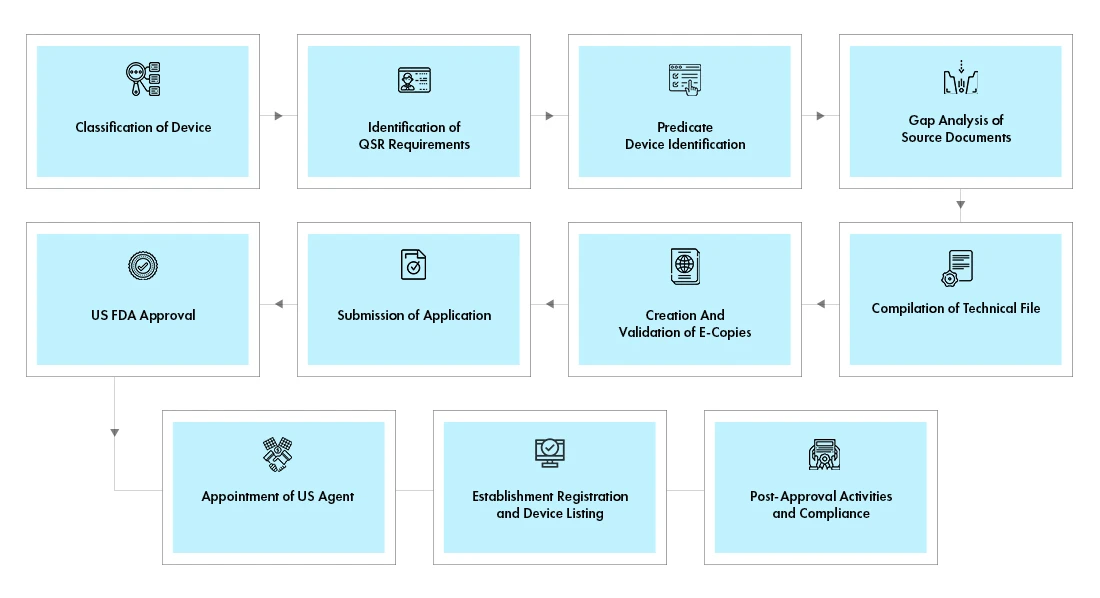
Management des Lebenszyklus von Geräten nach der Zulassung
Freyr ausländische Hersteller beim end-to-end Lebenszyklusmanagement end-to-end , einschließlich Aktivitäten nach der Zulassung, wie zum Beispiel:
- Änderungsmanagement nach der Zulassung - Änderungen bestehender Zulassungen für Medizinprodukte, z. B. Hinzufügung neuer Varianten und Zubehörteile, Hinzufügung neuer Anwendungsgebiete usw.
- Aufrechterhaltung von Zulassungen und Registrierung durch rechtzeitige Zahlung der MDUFA-Gebühren an die FDA
- Verbindungsstelle zwischen der US FDA dem Hersteller
Freyr verfügt über ein exklusives Lieferzentrum in den USA mit einem professionellen Team, das die Hersteller bei der Einhaltung der für die Zulassung erforderlichen Qualität und Sicherheit unterstützt. Die Intelligenzexperten von Freyrbeobachten aufmerksam die Aktualisierungen der Vorschriften und halten die Kunden über die Schritte auf dem Laufenden, die für die Konformität des Produkts mit dem aktuellen Standard erforderlich sind.
Zusammenfassung
| Risiko | Geräteklasse | QMS-Audit | Prädikat Verfügbarkeit | Regulierungspfad | U.S. Agent | US FDA |
|---|---|---|---|---|---|---|
| Geringes Risiko | I | Nein | NA | Befreit | Ja | 1 Monat |
| Mittleres Risiko | II | Ja (nach der Genehmigung) | Ja | PMN/510(k) | Ja | 9 - 12 Monate |
| Mittleres Risiko | II | Ja (nach der Genehmigung) | Nein | Antrag auf De-Novo-Einstufung | Ja | 18 - 30 Monate |
| Hohes Risiko | III | Ja (vor der Genehmigung) | NA | PMA | Ja | 18 - 30 Monate |
FreyrDienstleistungen für die Registrierung von Medizinprodukten
Freyr-Podcasts
- Regulatorische Due-Diligence-Prüfung
- Gerätedokumentation
- 513(g) Unterstützung
- 510(k)-Registrierung
- De-Novo-Antrag auf Einstufung
- PMA-Registrierung
- 21 CFR 820 Einhaltung der Vorschriften
- BIMO Audit-Unterstützung
- MDSAP Einhaltung der Vorschriften
- Unterstützung bei der Etikettierung
- Unterstützung bei Veröffentlichung und Einreichung
- U.S. Agent
- Q-Submission Treffen
- RFD- und Pre-RFD-Sitzungen
- Zertifizierung für kleine Unternehmen
- Registrierung von Betrieben und Auflistung von Geräten
- Einhaltung von Vorschriften für medizinische Geräte mit Strahlung
- Änderungsmanagement nach der Genehmigung
- Post Market Surveillance
- UDI-Einhaltung
- Regulatorische Beratung zur Behebung von Mängeln