
5 Minuten lesen
Das Medical Device Single Audit Program MDSAP) ermöglicht es einer anerkannten Auditierungsorganisation (AO), eine einzige Auditierung des Quality Management System (QMS) eines Medizinprodukteherstellers durchzuführen. Es enthält relevante regulatorische Anforderungen für fünf Länder, nämlich Brasilien (ANVISA), die USA (FDA), Japan (PMDA), Kanada (Health Canada) und Australien (TGA). Neben den teilnehmenden Regulierungsbehörden sind mehrere andere internationale Partner (die offiziellen Beobachter und assoziierten Mitglieder) am MDSAP beteiligt.
MDSAP ist von Health Canada Produkte der Klassen II, III und IV vorgeschrieben, für die anderen vier Länder jedoch freiwillig. Sie hat die Transparenz und die Angleichung der Vorschriften zwischen den teilnehmenden Behörden gefördert und die Notwendigkeit mehrfacher Audits minimiert, wodurch Zeit und Ressourcen der Medizinproduktehersteller eingespart werden konnten. Um Ihnen einen besseren Überblick über das MDSAP zu geben, haben wir hier versucht, die fünfzehn (15) am häufigsten gestellten Fragen zu beantworten.
- Warum wurde MDSAP -Programm entwickelt, obwohl es bereits eine weltweit anerkannte ISO 13485 gibt?
MDSAP entwickelt, um den Aufwand für behördliche Audits für Hersteller von Medizinprodukten zu reduzieren und eine bessere Abstimmung der regulatorischen Ansätze und technischen Anforderungen auf der Grundlage internationaler Standards und bewährter Verfahren zu fördern. Der Schwerpunkt liegt auf der Schaffung von Konsistenz, Vorhersehbarkeit und Transparenz in Regulierungsprogrammen durch die Standardisierung von Verfahren und Praktiken von Regulierungsbehörden und externen Audit-Organisationen.
Die Prüfung basiert auf den QMS-Anforderungen gemäß ISO 13485 den behördlichen Anforderungen des teilnehmenden Landes, in dem die Medizinprodukte vermarktet werden sollen.
- Was sind die Zulassungskriterien für die Durchführung eines MDSAP ?
Jeder Hersteller von Medizinprodukten, der beabsichtigt, sein Produkt in den teilnehmenden Ländern zu vermarkten, kann sich einem MDSAP unterziehen. Jede Regulierungsbehörde kann jedoch bei Bedarf Ausschlusskriterien für bestimmte Bedingungen festlegen.
In Japan gibt es beispielsweise folgende Ausnahmen für die Förderfähigkeit:
- Eine registrierte Produktionsstätte (RMS), die Medizinprodukte aus menschlichem/tierischem Gewebe herstellt
- ein RMS, das radioaktive IVDs herstellt, und
- Einrichtung eines Inhabers der Genehmigung für das Inverkehrbringen (MAH)
- Umfasst das MDSAP auch Kombinationsprodukte?
Medizinprodukte, die Arzneimittel (medizinische Substanzen) oder Biologika (z. B. Materialien tierischen Ursprungs, die unwirksam gemacht wurden, oder Gewebe, Zellen oder Substanzen mikrobiellen oder rekombinanten Ursprungs, menschliches Blut oder Extrakte aus menschlichem Blut oder Blutprodukten usw.) enthalten , gelten als Kombinationsprodukte und können in den Umfang eines MDSAP einbezogen werden.
Aufgrund von Unterschieden in der Regulierung dieser Produkte in den Zuständigkeitsbereichen der beteiligten Regulierungsbehörden können MDSAP und Zertifizierungsdokumente jedoch in einigen Zuständigkeitsbereichen nicht als Alternative zu den Inspektions- und Bewertungsanforderungen angesehen werden.
Australien – Kombinationsprodukte unterliegen einer externen Prüfung durch die TGA im Rahmen der australischen Konformitätsbewertung. Eine wirksame MDSAP kann jedoch die Inspektionen für diese Produkte reduzieren.
Brasilien, Japan – Kombinationsprodukte, die als Medizinprodukte gelten, sind im MDSAP enthalten, da es keine spezifischen Anforderungen hinsichtlich des QMS gibt.
Kanada – MDSAP deckt die QMS-Anforderungen für Kombinationsprodukte ab, die als Medizinprodukte gelten.
USA – MDSAP gelten nicht als Alternative zu FDA für Kombinationsprodukte.
- Kann ich das Land auswählen, das für das MDSAP in Betracht kommt?
Ja, das Audit wird in dem Umfang durchgeführt, der im Antrag auf Zertifizierungsdienste angegeben wurde. Von Medizinprodukteherstellern wird erwartet, dass sie die Vorschriften nur in den Ländern einhalten, in denen ihre Produkte vermarktet werden sollen.
- Ich bin ein Medizinproduktehersteller aus den US und beabsichtige, mein Produkt ausschließlich in Japan zu vermarkten. Ich stehe kurz vor einem MDSAP . Muss ich auch die Anforderungen anderer Länder erfüllen?
Nein, Hersteller von Medizinprodukten müssen nur ISO 13485 und Vorschriften ISO 13485 in den Ländern erfüllen, in denen ihre Produkte vermarktet werden sollen.
- Meine Prüforganisation (AO) und meine Europäische Benannte Stelle sind dieselbe. Kann ich für beide gleichzeitig auditiert werden?
Wenn Ihre AO und Ihre europäische benannte Stelle identisch sind, kann die Konformitätsbewertung nach Durchführung des MDSAP erfolgen und muss nicht gleichzeitig durchgeführt werden. Europäische benannte Stellen sind Beobachter für MDSAP, und die Konformitätsbewertung wird gemäß der EU MDR durchgeführt. Für MDSAP erfolgt die Bewertung gemäß den Anforderungen der ISO 13485 den regulatorischen Anforderungen der teilnehmenden Länder im Geltungsbereich.
- Was ist der Unterschied zwischen Stufe I und II der Bewertung?
MDSAP MDSAP-Erstprüfungsprozess umfasst zwei Stufen. Die Erstprüfung, auch Erstzertifizierungsprüfung genannt, besteht aus einer Stufe-I- und einer Stufe-II-Prüfung.
Das Audit der Stufe I umfasst die Überprüfung der Dokumentation und die Bewertung der Bereitschaft des Medizinprodukteherstellers, sich einem Audit der Stufe II zu unterziehen.
Die Auditphase II wird durchgeführt, um zu überprüfen, ob alle geltenden Anforderungen der ISO 13485 andere regulatorische Anforderungen der zuständigen Aufsichtsbehörde umgesetzt wurden.
- Wie viele Auditoren kann ich für ein MDSAP erwarten?
Die Bestimmung der Auditzeit legt fest, wie die Auditdauer vor Ort in Manntagen zu bestimmen ist. Die AO entscheidet, wie viele Auditoren das Auditteam bilden werden. Zum Beispiel kann ein Audit von (06) Manntagen in drei (03) Tagen von einem Team von zwei (02) Auditoren abgeschlossen werden.
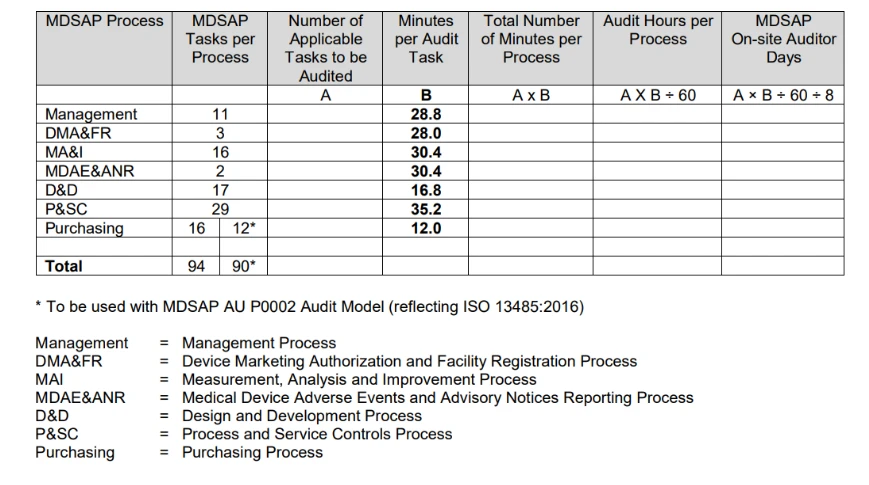
- Wie wird der Zeitpunkt für das MDSAP festgelegt?
Das von der FDA herausgegebene Verfahren zur Festlegung der Auditdauer fasst den Prozess zur Berechnung der Auditdauer in der folgenden Tabelle zusammen.
Die Berechnung der Auditdauer basiert in erster Linie auf der Anzahl der anwendbaren Auditaufgaben, die mit der Art des durchzuführenden Audits und den spezifischen Aktivitäten der zu auditierenden Organisation verbunden sind.
Ausführliche Informationen hierzu finden Sie unter MDSAP .
- Gibt es einen Leitfaden oder eine Checkliste, auf die ich zugreifen kann, um die Einhaltung eines MDSAP sicherzustellen?
Ja, Sie können auf das Dokument MDSAP Approach“ zugreifen. Es handelt sich um einen gut strukturierten Leitfaden der USFDA Querverweise zu bestimmten Abschnitten der ISO 13485:2016 und zu relevanten Vorschriften der australischen TGA, ANVISA brasilianischen ANVISA, der kanadischen Health Canada,PMDA japanischenPMDA und der US FDA enthält.
- Welche Rolle spielt ein Beobachter bei einem MDSAP ?
Ein MDSAP ist eine Aufsichtsbehörde, die an Sitzungen, Bewertungen und anderen Aktivitäten teilnehmen darf, aber die MDSAP nicht nutzt. Die Beobachter werden im MDSAP Authority Council (RAC) durch einen leitenden Manager vertreten.
- Was sind die nächsten Schritte, wenn ich eine Note von 4 oder höher erhalten habe?
Das Bewertungssystem wird für die während des Audits von der AO festgestellten Nichtkonformitäten vergeben. Eine Punktzahl von 4 oder 5 bedeutet ein hohes Risiko für ein Eingreifen. Sie müssen für jede festgestellte Nichtkonformität innerhalb von 15 Kalendertagen nach dem Datum des Berichts über die Nichtkonformität einen Abhilfeplan vorlegen. Der Abhilfeplan muss die Ergebnisse der Untersuchung der Nichtkonformität, ihre Ursachen und die geplanten Korrekturmaßnahmen zur Vermeidung eines erneuten Auftretens enthalten. Der Nachweis über die Umsetzung des Abhilfeplans/der Abhilfemaßnahmen sollte innerhalb von dreißig (30) Kalendertagen nach Abschluss des Audits erbracht werden.
- Gibt es einen Unterschied in der Herangehensweise an die Prüfung durch einen internen Prüfer im Vergleich zu einer AO?
MDSAP einen prozessorientierten Ansatz. Die AO wird wahrscheinlich Zusammenhänge und Verbindungen untersuchen, während ein interner Auditor sich eher auf jeweils einen funktionalen Aspekt konzentrieren wird. Daher kann es vorkommen, dass die AO eine Nichtkonformität in einem Funktionsbereich feststellt und Antworten in einem anderen Funktionsbereich sucht. Die Anwendung des prozessorientierten Ansatzes kann jedoch während einer internen Prüfung störend sein.
- Kann ich beim AO Einspruch einlegen, wenn ich beweisen kann, dass eine eingetragene Nichtübereinstimmung nicht gültig ist?
AO verfügt über ein Einspruchs- oder Anfechtungsverfahren, das Sie nutzen können, wenn Sie nachweisen können, dass eine aufgezeichnete Nichtkonformität ungültig ist. Allerdings können die den Nichtkonformitäten zugewiesenen Noten nicht aufgrund von Korrekturmaßnahmen geändert werden. Sie können nur auf der Grundlage von Beweisen geändert werden, die zeigen, dass sie ungültig waren.
- Wie lange ist das MDSAP gültig?
Medizinproduktehersteller, die im Rahmen des MDSAP zertifiziert sind, werden gemäß einem dreijährigen Zertifizierungszyklus jährlich auditiert. Das Erstaudit ist ein vollständiges Audit des QMS des Medizinprodukteherstellers. Darauf folgen zwei (02) aufeinanderfolgende Jahre mit jährlichen Überwachungsaudits. Der Zyklus beginnt im dritten Jahr mit einem Rezertifizierungsaudit von Neuem.
Um mehr über unsere MDSAP zu erfahren, wenden Sie sich noch heute an Freyr, um einen Termin für ein Gespräch mit unseren Experten zu vereinbaren.









