
2 Minuten lesen
Nachdem die USFDA erstmals Nitrosamin-Verunreinigungen in Arzneimitteln und pharmazeutischen Wirkstoffen (APIs) entdeckt hatte, schlossen sich auch die EU-Regulierungsbehörden vielen anderen Ländern an, um die damit verbundenen Risiken zu vermeiden. Sie haben mehrere Medikamente zurückgerufen, die aufgrund dieser Substanz Gesundheitsrisiken bergen. Nitrosamin ist für seine krebserregenden Eigenschaften bekannt und kann sich als schädlich erweisen, wenn es in Mengen über den für Menschen akzeptierten Werten aufgenommen wird. Die Substanz N-Nitrosodimethylamin (NDMA) hat einen zulässigen Grenzwert von 96 ng/Tag, und alles, was darüber liegt, ist in Arzneimitteln und APIs unzulässig.
Nitrosamine entstehen, wenn sekundäre, tertiäre oder quaternäre Amine mit einem Nitrosierungsmittel reagieren. Alle Arzneimittel, deren Wirkstoffe chemisch synthetisiert wurden, werden auf Verunreinigungen überprüft. Der Ausschuss für Humanarzneimittel (CHMP) hat einen Bewertungsbericht geprüft und erstellt. Er hat die Inhaber von Genehmigungen für das Inverkehrbringen (MAHs) aufgefordert, die neuesten Leitlinien für die Überprüfung aller derzeit auf dem Markt erhältlichen chemischen und biologischen Arzneimittel für den menschlichen Gebrauch zu befolgen.
MAHs sind dafür verantwortlich, dass der Herstellungsprozess aller biologischen und chemischen Produkte regelmäßig überprüft wird, um etwaige Verunreinigungen festzustellen. Sobald solche Verunreinigungen festgestellt werden, müssen geeignete Maßnahmen ergriffen werden, um die damit verbundenen Risiken zu mindern. Obwohl die Wahrscheinlichkeit solcher Verunreinigungen bei der Herstellung chemischer und biologischer Arzneimittel geringer ist, geht die Europäische Arzneimittelagentur (EMA) kein Risiko ein. Pharmazeutische Unternehmen müssen sicherstellen, dass die entsprechenden Herstellungsprotokolle gemäß den neuesten EMA vorhanden sind. Sie sind auch dafür verantwortlich, den Nitrosamingehalt in den auf dem Markt erhältlichen Arzneimitteln zu überprüfen und innerhalb der zulässigen Grenzwerte zu halten.
Nach Abschluss einer Überprüfung gemäß Artikel 5 Absatz 3 der Verordnung (EG) Nr. 726/2004 (Aufforderung zur Überprüfung) im letzten Jahr EMA die EMA neue Leitlinien zur Vermeidung von Nitrosaminverunreinigungen in Humanarzneimitteln herausgegeben. Das Verfahren ist für national zugelassene und zentral zugelassene Produkte identisch. Die EMA wird zusammen mit der Europäischen Direktion für die Qualität von Arzneimitteln und Gesundheitsfürsorge (EDQM) den CHMP-Artikel 5 Absatz 3 umsetzen. Hier ist das Verfahren, das befolgt werden sollte, um die Änderungen einzuhalten.
Der dreistufige Leitfaden für MAHs
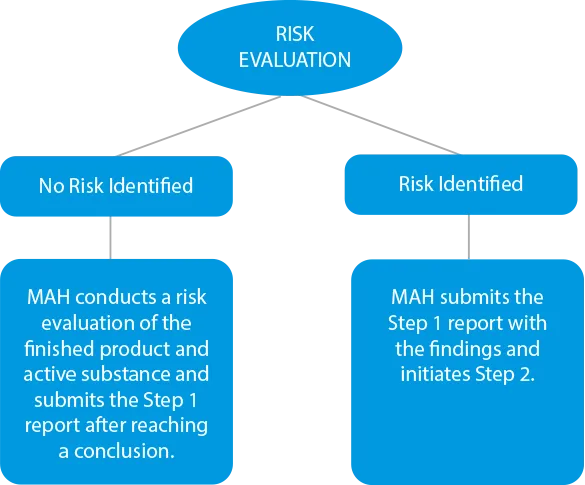
- Risikobewertung - Die Hersteller müssen ein Risikobewertungsverfahren durchführen, um die Wirkstoffe und das Endprodukt zu identifizieren und den Nitrosamingehalt zu überprüfen. Falls es Fälle von Kreuzkontamination gibt, sollte dies ebenfalls in den Ergebnisbericht aufgenommen werden. Die Fristen für die Einreichung in dieser Phase wurden für chemische Arzneimittel auf den 31. März 2021 und für biologische Arzneimittel auf den 01. Juli 2021 festgesetzt.
![]()
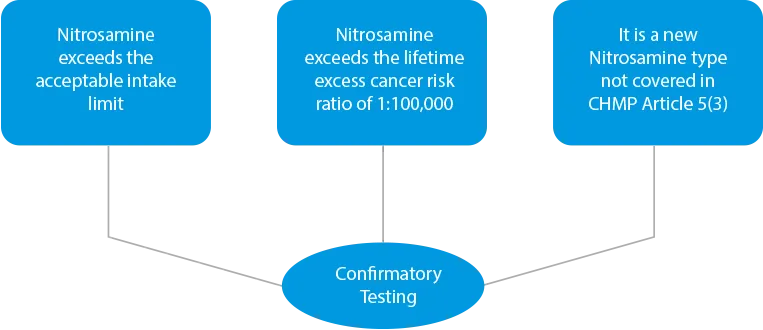
- Bestätigende Tests - Werden Kreuzkontaminationen festgestellt oder werden Produkte aufgrund höherer Nitrosamingehalte als riskant eingestuft, müssen Bestätigungstests durchgeführt werden. Bestätigungstests sind in drei (03) Fällen vorgeschrieben.
![]()
- Änderungen der Marktzulassung – Wenn Nitrosamin nachgewiesen wird, müssen zwei (02) Bestätigungstests durchgeführt werden, um der EMA die korrekten Messwerte zu melden. Auf dieser Grundlage müssen die Zulassungsinhaber eine Änderung des Herstellungsprozesses beantragen. Dies erfolgt unter Verwendung der standardmäßigen Regulierungsverfahren mit Hilfe einer Änderung der Marktzulassungsvorlage. Die Fristen hierfür sind der 26. September 2022 für chemische Produkte und der 1. Juli 2023 für biologische Arzneimittel.
Der ideale Weg für Hersteller von Arzneimitteln zur Einhaltung der neuen Nitrosaminwerte
Da dies die neueste Entwicklung ist, die die EMA vorgeschlagen EMA , um die Risiken im Zusammenhang mit Nitrosamingehalt in Arzneimitteln und Kreuzkontaminationen zu mindern, kann der gesamte Prozess für Zulassungsinhaber und Wirkstoffhersteller ziemlich überwältigend sein. Ob es nun darum geht, die Antwortvorlage einzureichen, wenn in Schritt 1 eine Kontamination festgestellt wird, oder Folgeuntersuchungen durchzuführen – jede Phase muss den neuen Vorschriften entsprechen. MAHs müssen mit Regulierungsexperten zusammenarbeiten, die über alle aktuellen Änderungen auf dem Laufenden sind und die Einhaltung der neuen Leitlinien sicherstellen. Wählen Sie den richtigen Partner wie Freyr Verzögerungen und Fehler zu vermeiden.









