
4 Minuten lesen
EINHALTUNG DER IDMP FRIST BIS JULI 2016: KEINE TRIVIALE AUFGABE
Es gibt viele Fragen zu IDMP B.: Wo befinden sich die Daten in einem Unternehmen? Ist eine Bereinigung und Angleichung der aktuellen Daten erforderlich? Wie organisieren Unternehmen diese Daten, damit sie leicht abgerufen und übermittelt werden können? Verfügt das Unternehmen über Prozesse, die von einem zentralen Repository profitieren können?
Die Einhaltung IDMP(Identification of Medicinal Products) ist keine triviale Angelegenheit, da dabei verschiedene regulatorische und betriebliche Zwänge berücksichtigt werden müssen. Unternehmen müssen warten, bis die Behörden endgültige Leitlinien herausgeben und Spezifikationen genehmigen. Gleichzeitig bleibt ihnen durch das Warten auf diese Leitlinien möglicherweise nicht genügend Zeit, um ihre Angelegenheiten in Ordnung zu bringen.
IDMP ein komplexer Standard mit weitreichenden Auswirkungen auf Daten, der die Zusammenarbeit und co vieler funktionsübergreifender Einheiten erfordert. Der Übergang bietet die Möglichkeit, die Geschäftsprozesse und IT-Fähigkeiten einer Organisation über viele Funktionseinheiten hinweg end-to-end . Außerdem trägt er dazu bei, ein robustes Change-Management-System zu etablieren.
Unternehmen müssen sich bewusst sein, dass eine gute Informationsarchitektur viel Aufwand und Zeit erfordert, und sie müssen erkennen, dass IDMP nicht einfach nur eine größere XEVMPD IDMP . Es ist auch wichtig zu beachten, dass ICH nur einen Teil des Ganzen ausmachen und dass regionale Leitlinien von entscheidender Bedeutung sind. Man muss auch berücksichtigen, dass parallele regionale Implementierungen unterschiedliche Reichweiten und Zeitrahmen haben werden und eine Vielzahl von Datenlieferanten umfassen.
Darüber hinaus müssen Pharmaunternehmen, die in regulierten Regionen vermarkten möchten, ab 2016 IDMP sein. Vor kurzem EMA IDMP und stellte den IDMP Stand IDMP sowie den Zeitplan vor. Nach Gesprächen mit der Pharmaindustrie und Softwareanbietern sowie einer Analyse der Verfügbarkeit ihrer eigenen Systeme und Ressourcen EMA geplant, die gesamteIDMP in mehrere Phasen zu unterteilen. Dieser Plan wird der European Commission EK) zur Genehmigung vorgelegt. Wenn die EMA die EK überzeugen kann, wird IDMP in der EU zwischen 2016 und 2018 erfolgen. Im schlimmsten Fall, wenn die EK nicht zustimmt, gibt es keinen Plan B. Die Strafen für die Nichteinhaltung betragen bis zu 5 % des Umsatzes eines Unternehmens. Keine gute Lösung für IDMP anfängliche und fortlaufende IDMP zu haben, ist ein Risiko, das sich kein Pharmaunternehmen leisten kann.
IDMP: ALS GLOBALE NORMENREIHE NACH ISO ENTWICKELT
Sollten in einem der ISO-Länder Rechtsvorschriften zur Identifizierung von Arzneimitteln eingeführt werden müssen, so erfolgt dies auf der Grundlage IDMP . Sobald verschiedene Regionen die IDMP übernommen haben, wird die Dateneingabe einheitlich sein, und globale Unternehmen und Regulierungsbehörden werden Zugang zu Daten haben, um die Konsistenz zwischen den Regionen zu überprüfen. Darüber hinaus werden gemeinsame kontrollierte Vokabulare diesen Prozess erheblich erleichtern.
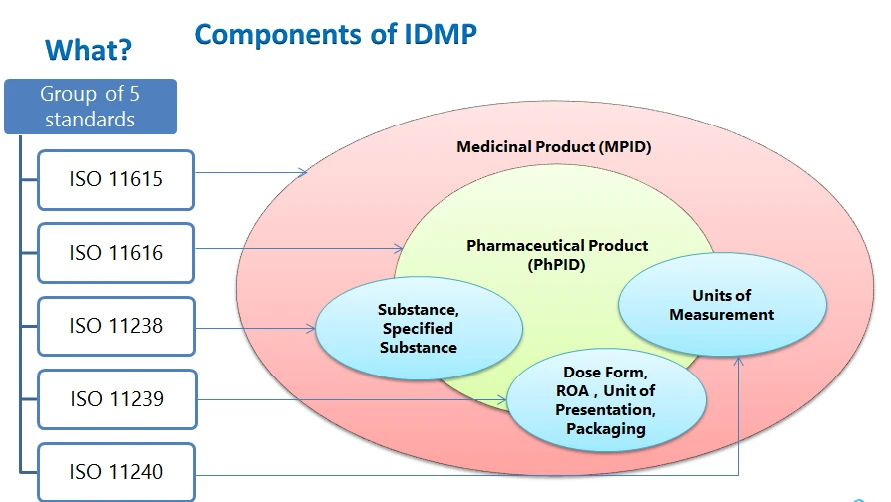
IDMP den strukturellen Elementen von IDMP die Arzneimittel-ID, die pharmazeutische Produkt-ID, die Substanz-ID, kontrollierte Vokabulare – Darreichungsform, Verabreichungsweg, Verpackungskomponenten, Darreichungsformen, Maßeinheiten und die Beschreibung des Herstellungsprozesses, die einzureichen sind.
IDMP
- ISO 11615 - Informationen zu Arzneimitteln
- ISO 11616 - Informationen über pharmazeutische Produkte
- ISO 11238 - Stoffe
- ISO 11239 - Pharmazeutische Darreichungsformen, Darreichungseinheiten, Verabreichungswege und Verpackung
- ISO 11240 - Maßeinheiten
AKTUELLER STATUS
ICH Implementierungsleitfäden und überarbeitet ISO-Normen.
EU-REGULATOREN BEREITEN SICH AUF IDMP VOR
- EMA die Zusammenarbeit mit dem EU-Regulierungsnetzwerk aufgenommen, um die Geschäftsmodelle für die IDMP zu definieren.
- Die EMA eine „EU-ISO IDMP zu diesem Zweck Experten aus EMA und dem EU-Netzwerkdatenrat rekrutieren. Konkret wird sie:
- Definieren Sie obligatorische und optionale ISO IDMP elemente.
- Definieren Sie Geschäftsregeln für optionale Datenelemente
- Definition von Konformität und Datentypen
- Definition der EU-Governance-Modelle
STANDPUNKT US
- Aktive Führungsrolle bei der IDMP in Zusammenarbeit mit den globalen (ehemaligen ICH)Regulierungsbehörden und der ISO
ANDERE REGULIERUNGSBEHÖRDEN
- Schweiz - Beabsichtigt die Umsetzung nach der EU (Fast Follower)
- Japan und Kanada haben Regulierungsbehörden als Sachverständige für die Gruppe Substance IG benannt
TIMELINES
EUROPÄISCHE EINFÜHRUNGSLEITFÄDEN
- Entwurf von Umsetzungsleitfäden eingeleitet und Leitfäden werden ab Q1 2016 verfügbar sein
SONSTIGES
- FDA noch FDA Datum, beabsichtigt jedoch, SPL nach Bedarf weiterzuentwickeln
- Japan – Unsicher in Bezug auf ICH , arbeitet ICH derzeit innerhalb der Regulierungsgruppe und der ISO
- Kanada - Wird voraussichtlich eingeführt, aber noch keine Einzelheiten
- Schweiz - Noch keine Updates
VERSTÄNDNIS DER IDMP
IDMP Informationen über Arzneimittel in Form einer Reihe von Standardkennungen, die auf einer Identifikationshierarchie basieren, die bei der Erstellung des EudraVigilance Medicinal Product Dictionary (EVMPD) oder in seiner erweiterten Form (xEVMPD) erstellt wurde. Es wird Überschneidungen mit den Informationen geben, die in Structured Product Labeling (SPL) in den US anderen Produktregistern weltweit.
IDMP jedoch IDMP neue Identifikatoren, neue Kategorien und neue Möglichkeiten, die Beziehungen zwischen Elementen im Datenmodell auszudrücken. IDMP in die DNA der Organisation integriert werden, da es die Erstellung von Datenmodellen im gesamten Unternehmen vorantreiben muss. Die IT-Infrastruktur einer Organisation kann es dann über mehrere Systeme, Geschäftsprozesse und Funktionseinheiten wie RA, Sicherheit, F&E, Dokumentation und Fertigungsprozesse hinweg erkennen.
HERAUSFORDERUNGEN BEI IDMP
Organisatorisches
- Auf mehrere Abteilungen verteilte Daten
- Seniorensponsoring zur Förderung der Teilnahme erforderlich
Technisch
- Entdecken, sammeln und konsolidieren Sie saubere Daten
- 250 bis 300 Felder pro Produkt
Co
- Management des kontinuierlichen Wandels
- Co zwischen mehreren Teams
- Aufrechterhaltung der Datenregulierung mit internen Verfahren
SCHLUSSFOLGERUNG:
IDMP: AUSWIRKUNGEN AUF DIE PHARMAINDUSTRIE
Die Umsetzung der IDMP wird sich voraussichtlich auf die Vorbereitung und Planung von Einreichungen sowie auf die Pflege unternehmensweiter Daten auswirken, darunter Fertigungsdaten und strukturierte Stoffinformationen für Registrierungszwecke.
IDMP : EFFIZIENZGEWINNE BEI DER UMSETZUNG GARANTIERT
Um sicherzustellen, dass Ihr Unternehmen für die IDMP gerüstet ist, ist eine enge Zusammenarbeit zwischen mehreren Abteilungen innerhalb Ihres Unternehmens erforderlich. Ein kompetenter Dienstleister mit einem exklusiven Portfolio an regulatorischen Kompetenzen kann Ihnen dabei helfen, IDMP zu erreichen, wodurch Ihr Unternehmen auf neue, sich entwickelnde Chancen auf dem Markt reagieren kann.









