2 Minuten lesen
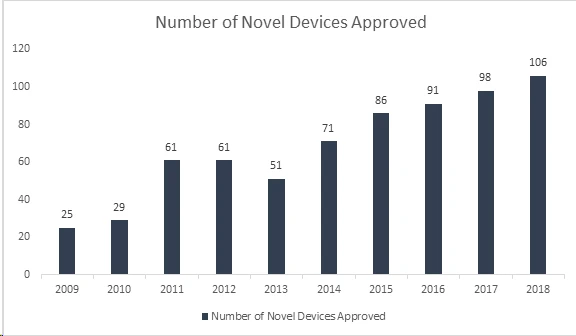
Wussten Sie schon? Die US and Drug Administration (FDA) hat mit der Zulassung von 106 neuartigen Medizinprodukten im Jahr 2018 einen weiteren Rekord aufgestellt und damit das erfolgreichste Jahr für Medizinprodukte erzielt. Mit dieser Leistung FDA ihren 40 Jahre alten Rekord aus dem Jahr 2017 von 99 zugelassenen neuartigen Medizinprodukten übertroffen und damit ein kontinuierliches Wachstum in den letzten 8 Jahren gezeigt. Zu den zugelassenen Produkten gehören eine Reihe innovativer Produkte wie automatisierte Insulindosierungssysteme für Kinder, die weltweit kleinste Herzklappe für Neugeborene, die erste mobile medizinische App zur Behandlung von Opioid- und Substanzmissbrauch sowie Technologien mit künstlicher Intelligenz zur Diagnose von diabetischer Retinopathie.
FDA stets die Sicherheit und Innovation von Medizinprodukten gefördert, um deren hohe Qualität zu gewährleisten. Um mit der steigenden Zahl von Zulassungen für neuartige Produkte und deren Sicherheit Schritt zu halten, FDA eine „Modernisierung” des Zulassungsverfahrens für Medizinprodukte. Nach Angaben der Behörde könnte die Modernisierung des Zulassungsverfahrens eine neue Befugnis erfordern. Im Jahr 2018 veröffentlichten FDA das Center for Devices and Radiological Health (CDRH) ein gemeinsames Dokument, in dem sie feststellten, dass 510(k) eine der beiden Arten von Anträgen ist, die zur Definition von neuartigen Produkten hinzugefügt wurden. Zusammen mit dieser Änderung wurden auch Humanitarian Device Exemptions (HDEs) in die Definition von neuartigen Produkten aufgenommen, nachdem das CDRH aufgrund des 21st Century Act Änderungen an seinem Programm für bahnbrechende Medizinprodukte vorgenommen hatte.
Der Vorschlag der FDA des CDRH ist eine Reaktion auf die potenzielle Notwendigkeit, neue Befugnisse zur Modernisierung des 510(k)-Verfahrens zu erlangen. Das Motiv des Vorschlags ist es, die Verwendung von Referenzprodukten, die älter als 10 Jahre sind, als „Substantially Equivalent“ (SE) zu begrenzen, um Innovationen zu fördern. Dies ist ein Fortschritt gegenüber den Leitlinien, die die Behörde im April 2018 veröffentlicht hat. Der Entwurf wurde von der FDA veröffentlicht, FDA die Ausweitung des Abbreviated 510(k)-Programms bei FDACDRH unter dem Titel „Safety and Performance-based Pathway” vorzuschlagen. Er wurde eingeführt, um die Belastung durch Bestimmungen für Medizinprodukte zu verringern. Der Ansatz zielt auch darauf ab, die Effizienz der Prüfung von 510(k)-Anträgen zu erhöhen und damit den Druck auf die Behörde zu verringern.
Hier sind einige der wichtigsten Punkte des Leitfadens:
- Der neue Weg bewertet die Sicherheit und Wirksamkeit der Produkte anhand festgelegter Sicherheitsstandards und Leistungskennzahlen
- Trotz der neuen Normen müssen die Geräte die bestehenden Normen erfüllen, um vermarktet werden zu können
- Moderne Technologie wird anhand moderner Standards getestet
- Der Ansatz wird einen stärkeren Wettbewerb um die Entwicklung von sichereren Geräten fördern
Die Zahl der zur Zulassung vorgelegten Medizinprodukte ist im Laufe der Jahre exponentiell gestiegen. Dies hat der FDA einen großen Spielraum gegeben FDA innovative Maßnahmen zur Verbesserung der Zulassungsverfahren FDA verabschieden und umzusetzen. Die Behörde ist von den Vorteilen dieses Vorschlags überzeugt, aber die Reaktion der Industrie muss noch entschlüsselt werden.
Da die FDA neue Leitfäden zur Verbesserung der Registrierung von Medizinprodukten herausgibt, müssen Hersteller von Medizinprodukten diese im Auge behalten und entsprechend handeln. Bleiben Sie auf dem Laufenden. Bleiben Sie konform.









