
2 Minuten lesen
Die US FDA einen Leitfaden veröffentlicht, der der Industrie und den Mitarbeitern der Gesundheitsbehörde (HA) dabei helfen soll, zu bestimmen, wann eine Softwareänderung an einem Medizinprodukt den Hersteller dazu verpflichtet, eine neue Vorab-Meldung (510(k)) FDA einzureichen und deren FDA einzuholen. Dieser Leitfaden soll die Vorhersehbarkeit, Konsistenz und Transparenz des Entscheidungsprozesses „Wann muss eine Meldung eingereicht werden?“ verbessern, indem er einen möglichst unkomplizierten Ansatz bietet und den regulatorischen Rahmen, die Richtlinien und Praktiken beschreibt, die einer solchen Entscheidung zugrunde liegen, insbesondere in Bezug auf Softwareänderungen. Sehen wir uns den FDA im Detail an.
Leitprinzipien und Flussdiagramm FDA
Um den Herstellern von Medizinprodukten bei der Anwendung der wichtigsten Grundsätze zu helfen, enthält das Dokument ein Flussdiagramm, zusätzliche Erläuterungen und Beispiele, die für Entscheidungen über eine neue Premarket Notification 510(k) für eine Softwareänderung an einem bereits in den US zugelassenen Produkt erforderlich sind. Darüber hinaus sollten bei der Anwendung dieser Leitlinien mehrere Grundsätze beachtet werden, um zu entscheiden, ob eine neue 510(k) zur Änderung eines bestehenden Produkts eingereicht werden muss. Einige davon sind allgemein bekannt und leiten sich aus der aktuellen FDA (k) ab, andere sind für die Anwendung des in dieser Leitlinie erwähnten Logikschemas erforderlich. Gemäß der Leitlinie kann das bereitgestellte Schema nicht alle möglichen Feinheiten im Zusammenhang mit solchen Änderungen und deren Auswirkungen auf die Entscheidung abdecken. Um die Notwendigkeit einer neuen Premarket Notification 510(k) zu bestimmen, sollten Hersteller von Medizinprodukten daher die allgemeinen Grundsätze und das Flussdiagramm berücksichtigen, die im Folgenden zusammengefasst sind.
- Änderungen, die die Sicherheit oder Wirksamkeit eines Produkts beeinträchtigen
- Erste risikobasierte Bewertung
- Unbeabsichtigte Folgen von Veränderungen
- Einsatz des Risikomanagements
- Rolle der Prüfung (Verifizierungs- und Validierungsaktivitäten) bei der Bewertung, ob eine Änderung die Sicherheit und Wirksamkeit erheblich beeinträchtigen könnte
- Bewertung gleichzeitiger Änderungen, um festzustellen, ob die Einreichung einer neuen 510(k) erforderlich ist
- Geeignete Vergleichsinstrumente und die kumulative Wirkung von Änderungen
- Dokumentationspflicht (21 CFR Teil 820)
- 510(k)-Anträge für modifizierte Produkte
- Feststellung der substanziellen Gleichwertigkeit
- Die Einreichung einer neuen 510(k)-Meldung ist wahrscheinlich erforderlich, wenn ein Hersteller sein Produkt so verändert, dass die Sicherheit oder Wirksamkeit des Produkts beeinträchtigt wird. Änderungen, die sich nicht auf die Sicherheit oder Wirksamkeit des Produkts auswirken sollen, sollten jedoch trotzdem bewertet werden.
- Um festzustellen, ob eine Änderung oder Modifikation die Sicherheit oder Wirksamkeit erheblich beeinträchtigen könnte, sollte der Hersteller zunächst eine risikobasierte Bewertung durchführen, um festzustellen, ob die Änderung die Sicherheit oder Wirksamkeit des Produkts entweder positiv oder negativ beeinflussen könnte. Bei dieser risikobasierten Bewertung sollten alle neuen Risiken und Änderungen bestehender Risiken, die sich aus der Produktänderung ergeben, identifiziert und analysiert werden, um eine erste Entscheidung darüber zu treffen, ob eine neue 510(k)-Anmeldung erforderlich ist.
- Manchmal gibt es zusätzliche unbeabsichtigte oder ungeplante Folgen, die bei der Einreichung von Software ausgelöst werden können. Das Flussdiagramm sollte diese Folgen bewerten, um festzustellen, ob die Einreichung einer neuen 510(k) erforderlich ist.
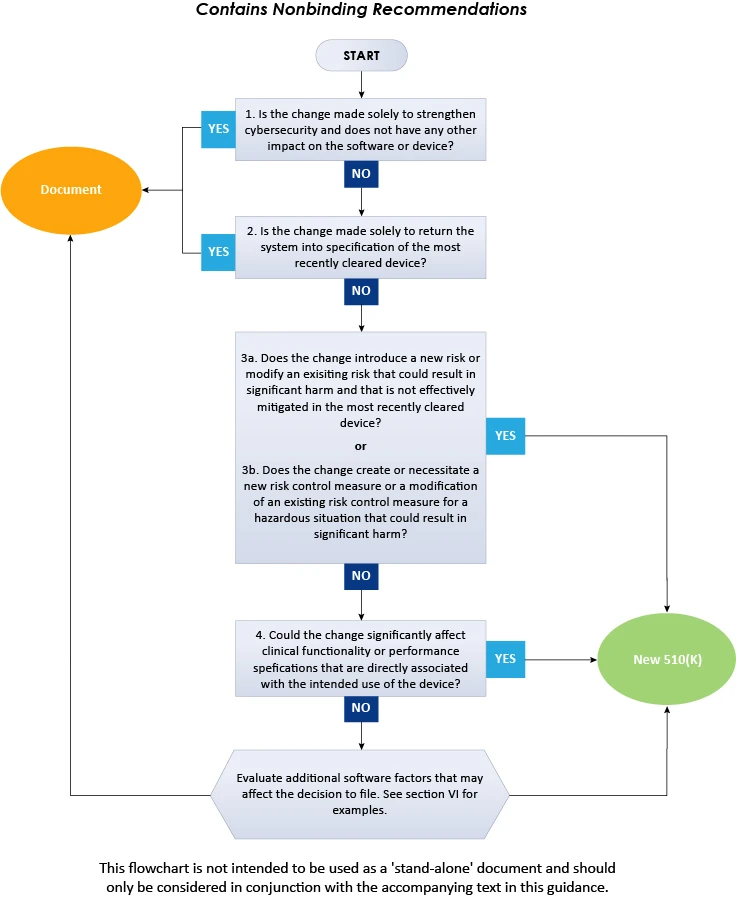
Das obige Flussdiagramm veranschaulicht ein schrittweises Verfahren, das bei der Entscheidung über die Einreichung eines 510(k)-Antrags für Softwareänderungen an bestehenden Geräten zu befolgen ist. Zusammenfassend lässt sich sagen, dass die vorliegende FDA detailliert beschreibt, wie Medizinproduktehersteller vorgehen müssen, wenn sie entscheiden, ob die Softwareänderungen an einem bestehenden Medizinprodukt die Einreichung eines neuen 510(k)-Antrags erfordern. Weitere Einblicke in die FDA erhalten Sie bei Freyr – einem bewährten Experten für regulatorische Fragen. Bleiben Sie informiert. Bleiben Sie konform.









