5 Minuten lesen
Einreichungen sind für den 24. September 2016 geplant.
Nachdem nun die zweite Phase der UDI-Konformität für Medizinprodukte der Klassen III I/LS/LS umgesetzt wurde, fragen sich viele Hersteller von Medizinprodukten, insbesondere der Klasse II, wie sie sich am besten auf die Frist für die Einreichung von Daten zu Produkten der Klasse II am 24. September 2016 vorbereiten können. Um ihnen einen Vorsprung zu verschaffen, haben wir bei Freyr einige Voraussetzungen identifiziert, die sie berücksichtigen sollten, um Produkte der Klasse II für die Einhaltung der UDI-Vorgaben FDAvorzubereiten.
Die neue Verordnung schreibt vor, dass alle Medizinprodukte der Klasse II mit einer eindeutigen Produktkennung (UDI) gekennzeichnet und verpackt sowie in die Global Unique Device Identification Database (GUDID) FDAaufgenommen werden müssen. Angesichts der Volatilität der Compliance-Anforderungen in Verbindung mit kürzeren Einreichungsfristen besteht die Herausforderung für Gerätehersteller darin, sich mit den Einzelheiten der Compliance-Prozesse vertraut zu machen. Gleichzeitig müssen sie sicherstellen, dass keine der wichtigen Geräteeigenschaften übersehen wird, während sie die über verschiedene Systeme verstreuten Gerätedaten zusammenführen und in Tabellenkalkulationen abgleichen, um Compliance-Berichte zu erstellen.
Um Geräteherstellern dabei zu helfen, diesen zeitkritischen und komplexen Konformitätsprozess fehlerfrei zu durchlaufen, Freyr die folgenden Voraussetzungen zusammengestellt, die zu beachten sind.
Bestimmen Sie das Datum für die Einhaltung der UDI-Vorschriften: Seit der Veröffentlichung der endgültigen Regelung FDA einige der Termine für die Einhaltung der Vorschriften für Medizinprodukte geändert und verlängert. Um die Strategien und Prozesse zur Einhaltung der Vorschriften sorgfältig im Voraus zu planen und übereilte Änderungen in letzter Minute zu vermeiden, sollten Etikettierer das genaue Datum für die Einhaltung der Vorschriften festlegen.
Geräte der Klasse II Datum der Einhaltung Compliance-Anforderungen 24. September 2016Produkte der Klasse III, die mit einer UDI gekennzeichnet werden müssen, müssen eine UDI als dauerhafte Kennzeichnung auf dem Produkt selbst tragen, wenn es sich um ein Produkt handelt, das dazu bestimmt ist, mehr als einmal verwendet zu werden und vor jeder Verwendung wieder aufbereitet zu werden. Die Etiketten und Verpackungen von Medizinprodukten der Klasse II müssen mit einer UDI Die Datumsangaben auf den Etiketten dieser Geräte müssen wie vorgeschrieben formatiert werden Eigenständige Software der Klasse II muss ihre UDI wie vorgeschrieben bereitstellen Daten für Produkte der Klasse II, die mit einer UDI gekennzeichnet werden müssen, müssen an die GUDID-Datenbank übermittelt werden Für die meisten Produkte ist das Konformitätsdatum für die direkte Kennzeichnung ein anderes als für die anderen Anforderungen. Je nachdem, ob Ihr Produkt zur Wiederverwendung oder zur Wiederaufbereitung bestimmt ist, bestimmen Sie das UDI-Konformitätsdatum für die Direktkennzeichnung wie hier gezeigt:
Direkte Kennzeichnung: Termine für die Einhaltung der Vorschriften Kategorie des Geräts - Wiederverwendet und wiederaufbereitet 24. September 2015 Lebenserhaltende und lebensunterstützende Geräte, unabhängig von der Geräteklasse 24. September 2016 Produkte der Klasse III und Produkte, die nach dem Public Health Service Act zugelassen sind Sep 24 2018 Produkte der Klasse II 24. September 2020 Produkte der Klasse I und nicht klassifizierte Produkte Prüfen Sie die Notwendigkeit einer direkten Kennzeichnung der UDI-Nummer: Alle Medizinprodukte, die mehr als einmal verwendet werden oder die vor jeder Verwendung aufbereitet werden müssen, müssen mit einer direkten Kennzeichnung der UDI-Nummer versehen werden. Eine Ausnahme bilden implantierbare Produkte, die gemäß der UDI-Vorschrift nicht direkt gekennzeichnet werden müssen. Produkte für den einmaligen Gebrauch, auch wenn sie wiederaufbereitet werden, müssen ebenfalls keine dauerhafte UDI tragen - 21 CFR 801.45(d)(3). Prüfen Sie daher die Notwendigkeit einer direkten Kennzeichnung auf der Grundlage der Kategorie der von Ihnen hergestellten Medizinprodukte.
- Plan für umfassende Einhaltung der VorschriftenÜberprüfen Sie die FDA für Ihre spezifischen Produkte, die erfüllt werden müssen. Führen Sie eine gründliche Lückenanalyse durch, um Defizite in Bezug auf Daten oder Technologien aufzudecken, die bei der Einhaltung der strengen FDA zu bewältigen sind. Zu den Herausforderungen können beispielsweise die Beschaffung von DI- oder PI-Informationen und die Verarbeitung großer Mengen unstrukturierter Daten aus unterschiedlichen Quellen gehören. Anstatt bis spät in die Nacht zu arbeiten, um alle Daten zu Medizinprodukten rechtzeitig abzugleichen, sollten Sie im Voraus eine umfassende Compliance planen und validierte Systeme und Tools einsetzen, die die Datenintegration, Datenqualität und Datenverwaltung unterstützen.
![]()
Erhalten Sie die DI-Nummer und die Mitgliedschaft in der Agentur: Die UDI setzt sich zusammen aus der Geräteidentifikation (DI – eindeutige Nummer basierend auf der Version oder dem Modell des Geräts) und der Produktidentifikation (PI – umfasst Chargennummer, Seriennummer oder Verfallsdatum). Der DI-Teil der UDI dient als Primärschlüssel für die Suche nach Informationen über das Gerät in der GUDID. Für die Vergabe der DI FDA die FDA drei ausstellende Stellen akkreditiert: GS1, HIBCC und ICCBBA. In diesem Szenario müssen Etikettierer Mitglied einer dieser Stellen werden, um die DI-Nummer zu erhalten, die in die GUDID FDAeingegeben werden muss.
![]()
Kennung Attribute Ausstellende Agenturen UDI DI (Device Identifier - Static Data)
Muss mit GUDID synchronisiert werdenEinzigartige Anzahl von
Hersteller
Gerät Marke
Gerät Modell
GSI
HIBCC
ICCBBAPI (Product Identifier - Dynamic Data)
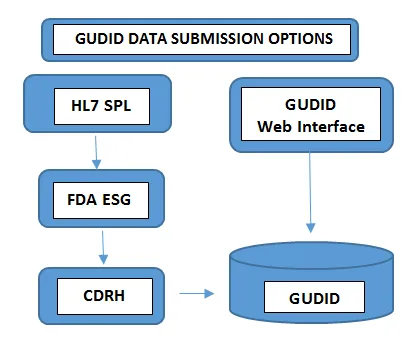
Erforderlich auf allen VerpackungsebenenLosnummer, Seriennummer, Herstellungsdatum Verfallsdatum, - Übermittlung der DatenDie Art und Weise, wie Sie die Daten an die GUDID übermitteln, hängt vom Umfang Ihres Produktportfolios ab. Gerätehersteller mit einer geringen Anzahl von Geräten entscheiden sich dafür, die UDI-Informationen manuell über die kostenlose GUDID-Webschnittstelle FDAzu übermitteln. In diesem Fall kann jeweils nur ein DI-Datensatz über eine gesicherte GUDID-Webschnittstelle übermittelt werden.Hersteller mit einem größeren Produktportfolio entscheiden sich hingegen für die HL7-SPL-Übermittlungsoption, um Daten elektronisch zu erfassen und die konsolidierten Daten vor der Übermittlung an das Electronic Submission Gateway (ESG) FDAunter Verwendung der DUNS-Nummer in das SPL-Format zu konvertieren. Bitte beachten Sie, dass das GUDID-Konto nicht nach Übermittlungsart unterschieden wird. Das Konto dient dazu, Sie als Etikettierer zu identifizieren, damit Sie Gerätedaten über beide Optionen übermitteln können.
![]()
- Einrichten eines GUDID-Kontos: Ein Etikettierer/Gerätehersteller benötigt je nach Anzahl der zuzuweisenden Rollen ein oder mehrere GUDID-Konten, darunter beispielsweise GUDID-Koordinator, Dateneingabe-Benutzer usw. Um jedoch jede Rolle für die Dateneingabe zu autorisieren, benötigt der Hersteller FDA der Kontoerstellung eine Genehmigung der FDA . Um ein entsprechendes GUDID-Konto zu erstellen, muss eine E-Mail-Anfrage an FDA gesendet werden. FDA erhält der Antragsteller, also Sie, ein Dokument zur Kontoanforderung, das ausgefüllt werden muss. Sobald Sie das ausgefüllte Dokument FDA E-Mail an die FDA zurücksenden, prüft die Behörde das Formular und sendet Ihnen anschließend eine E-Mail mit den Anmeldedaten für Ihr GUDID-Konto.


Die Umsetzung der UDI ist ein komplexer und zeitaufwändiger Prozess. Während des Kurses sehen sich Hersteller von Medizinprodukten bei der Erfüllung der UDI-Anforderungen FDAmit vielen Herausforderungen in Bezug auf Datenmanagement, Datenintegration und Datenübermittlung konfrontiert. Da die Frist für die Einhaltung der Vorschriften für Produkte der Klasse II nur noch ein Jahr entfernt ist, Freyr den Unternehmen, sich schon jetzt darauf vorzubereiten.
Um Ihr Unternehmen durch diesen komplexen Compliance-Prozess zu führen, Freyr das Beste aus beiden Welten – die bedarfsgerechte, vollständig konfigurierbare UDI-SoftwarelösungFreyr sowie ein Kompetenzzentrum (CoE of Excellence,CoE), das erstklassige, kostengünstige und anpassbareUDI-Dienstleistungenanbietet, die auf Ihre individuellen und anspruchsvollen Anforderungen zugeschnitten sind.









