
3 Minuten lesen
510(k) ist ein Antrag, der vor der Markteinführung bei der FDA eingereicht wird, FDA nachzuweisen, dass das zu vermarktende Produkt ebenso sicher und wirksam ist wie ein bereits legal vermarktetes Produkt (Referenzprodukt), d. h. im Wesentlichen gleichwertig ist. Produkte mit mittlerem Risiko müssen einen 510(k)-Antrag einreichen, darunter eine Minderheit der Produkte der Klassen I und III und eine Mehrheit der Produkte der Klasse II.
Es gibt drei (03) Arten von 510(k)-Programmen: herkömmliche, abgekürzte und spezielle. Der Sicherheits- und Leistungspfad wurde 2019 eingeführt und baut auf dem abgekürzten Programm auf. Das im Jahr 2020 eingeführte eSTAR-Programm ermöglicht eine umfassende Einreichung von Medizinprodukten über ein interaktives PDF-Formular.
Wer benötigt eine 510(k)-Zertifizierung?
510(k) ist im Wesentlichen die Bezeichnung für das Verfahren/den Weg, den Hersteller von Medizinprodukten, die ihre Produkte mit mittlerem bis hohem Risiko in den US vermarkten möchten, US , um nachzuweisen, dass das zu vermarktende Produkt genauso sicher und wirksam ist wie ein gesetzlich zugelassenes Produkt.
Im Folgenden wird das schrittweise Verfahren zur Erlangung einer 510(k)-Zulassung beschrieben.
Schritt 1 - Identifizierung des Geräteklassencodes, der Einreichungsart und des Prädikatsgeräts
- Produktcode und Regulierungsnummer identifizieren – Um die 510(k)-Prüfungsanforderungen zu bestimmen, müssen zunächst der Produktcode und die Regulierungsnummer identifiziert werden. Man kann eine Suche in der FDA starten, um die 7-stellige Regulierungsnummer zu finden, deren Identifizierung mit dem Verwendungszweck des betreffenden Produkts übereinstimmt.
- Der FDA besteht aus drei (03) Buchstaben. Anhand dieses Codes lassen sich Informationen zur Produktklassifizierung, zur Beschreibung der Vorschriften und zu den GMP-Anforderungen abrufen.
- Auswahl des Einreichungstyps – Ein Einreicher kann einen der drei (03) zuvor genannten Einreichungstypen wählen. Traditional 510(k) ist für Ersteinreichungen vorgesehen, Special 510(k) für Hersteller von Medizinprodukten, die Änderungen an einem bestehenden Produkt einreichen möchten, und Abbreviated 510(k) kann gewählt werden, wenn das Produkt den festgelegten freiwilligen Konsensstandards entspricht. Im Falle eines Abbreviated 510(k) muss sich der Einreicher auf FDA stützen.
- Identifizierung des Prädikatsprodukts - Ein Hersteller von Medizinprodukten muss nachweisen, dass das Produkt, das er in Verkehr bringen will, dieselbe Zweckbestimmung und dieselben technischen Merkmale aufweist wie das rechtmäßig in Verkehr gebrachte Produkt, das auch als Prädikatsprodukt bezeichnet wird. Wenn es Unterschiede bei den technischen Merkmalen gibt, muss der Antragsteller nachweisen, dass diese Unterschiede keine Bedenken hinsichtlich der Sicherheit und Wirksamkeit aufwerfen.
Schritt 2 - Vorbereitung der 510(k)-Datei
Der nächste Schritt besteht darin, die 510(k)-Datei, die Leitlinien und die Informationen vorzubereiten, die auf der FDA verfügbar sind. Dazu gehören die Checkliste für die Annahme aller drei (03) Arten von 510(k)-Programmen und eine Microsite mit dem Titel „Content for 510(k)“, die unter anderem Informationen zu Angaben zu Anwendungsgebieten, zum Vergleich der wesentlichen Gleichwertigkeit und zur vorgeschlagenen Kennzeichnung sowie weitere nützliche Informationen enthält.
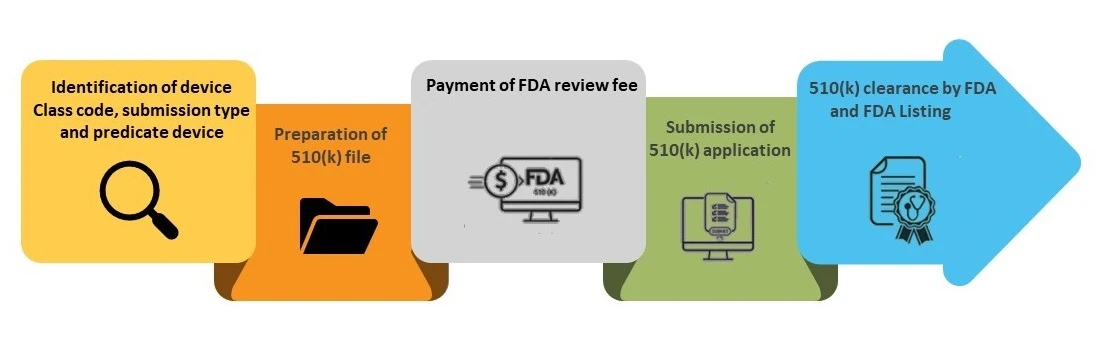
Schritte des 510(k)-Einreichungsverfahrens
Schritt 3 – Zahlung der FDA
Alle Arten von 510(k)-Anträgen sind gebührenpflichtig. Für das Haushaltsjahr 2023 beträgt die Standardgebühr für 510(k) 19.870 $. Für Unternehmen, die unter dem Centre for Diagnostics and Radiological Health (CDRH) zertifiziert sind, auch bekannt als Kleinunternehmen, beträgt die Gebühr 4.967 $. Die Gebühr kann sich im nächsten Haushaltsjahr ändern.
Schritt 4 - Einreichung des 510(k)-Antrags
Der Antragsteller kann über das CDRH-Portal eine elektronische Kopie (eCopy) oder eine elektronische Einreichungsvorlage und Ressource (eSTAR) für die Einreichung vor dem Inverkehrbringen senden.
Ab dem 01. Oktober 2023 müssen alle 510(k)-Anträge, sofern sie nicht gemäß den endgültigen Leitlinien ausgenommen sind, in elektronischer Form über eSTAR eingereicht werden.
Nach Einreichung des 510(k) wird eine eindeutige Kontrollnummer vergeben, die als „510(k)-Nummer” oder „K-Nummer” bezeichnet wird. FDA zwei Überprüfungen FDA : eine zur Überprüfung, ob die ordnungsgemäße Nutzungsgebühr entrichtet wurde, und eine zweite zur Überprüfung, ob eine gültige eCopy oder eSTAR vorgelegt wurde.
- Am Tag 07 FDA eine Bestätigungsmitteilung, sofern die ordnungsgemäße Nutzungsgebühr entrichtet und eine gültige eCopy oder eSTAR vorgelegt wurde. Andernfalls FDA die FDA ein Zurückhalteschreiben wegen ungelöster Probleme.
- Am 15. Tag FDA eine Abnahmeprüfung FDA . FDA dem Antragsteller FDA , ob der 510(k)-Antrag zur inhaltlichen Prüfung angenommen oder mit einem Ablehnungsbescheid (Refuse to Accept, RTA) zurückgestellt wird.
- Am 60. Tag FDA eine eingehende Prüfung FDA . FDA FDA über „
“ FDA , dass entweder eine interaktive Prüfung durchgeführt FDA oder das 510(k)-Verfahren ausgesetzt wird und zusätzliche Informationen angefordert werden.
Schritt 5 – FDA und Aufnahme in FDA (k)-Datenbank
Das Ziel der FDA ihre Entscheidung zu den Änderungen der Gebühren für Medizinprodukte (MDUFA) innerhalb von 90 FDA bekannt zu geben. FDA sind die Kalendertage zwischen dem Datum des Eingangs des 510(k)-Antrags und dem Datum der MDUFA-Entscheidung, wobei die Tage, an denen der Antrag aufgrund einer Aufforderung zur Einreichung zusätzlicher Informationen zurückgestellt wurde, nicht mitgezählt werden. MDUFA-Entscheidungen für 510(k)-Anträge umfassen Feststellungen zur wesentlichen Gleichwertigkeit (SE) oder Nicht-wesentlichen Gleichwertigkeit (NSE).
Wenn eine Entscheidung getroffen wurde, FDA dem Antragsteller per E-Mail ein Entscheidungsschreiben FDA . Ein 510(k)-Antrag, für den ein SE-Entscheidungsschreiben ausgestellt wurde, gilt als „genehmigt“. Er wird dann in der 510(k)-Datenbank zusammen mit den Indikationen für die Verwendung des Medizinprodukts und der 510(k)-Zusammenfassung oder der 510(k)-Erklärung als Anhänge aufgeführt.
Es lässt sich feststellen, dass eine sorgfältige Planung und Durchführung durch gründliche Dokumentation und ein tiefgreifendes Verständnis des regulatorischen Umfelds entscheidend für eine erfolgreiche Einreichung eines 510(k)-Antrags bei FDA sind.
Wenn Sie Hilfe beim Einreichungsprozess für Ihr Medizinprodukt gemäß 510(k) benötigen, schreiben Sie us bitte us sales@freyrsoltions.com oder vereinbaren Sie einen Termin für ein Telefonat mit unseren Experten, die Ihnen bei der Abwicklung der Verfahren behilflich sein können. Bleiben Sie auf dem Laufenden. Bleiben Sie konform.









