
3 Minuten lesen
Quality Management System (QMS) ein wesentlicher Bestandteil der Medizinprodukteindustrie und gewährleistet die Sicherheit, Wirksamkeit und Einhaltung der Vorschriften für Medizinprodukte während ihres gesamten Lebenszyklus. Das QMS wird in allen Phasen des Lebenszyklus von Medizinprodukten, einschließlich der Konstruktions- und Entwicklungsphase, implementiert, um sicherzustellen, dass das Produkt den gesetzlichen und den Anforderungen der Anwender entspricht und dass potenzielle Risiken identifiziert und behoben werden.
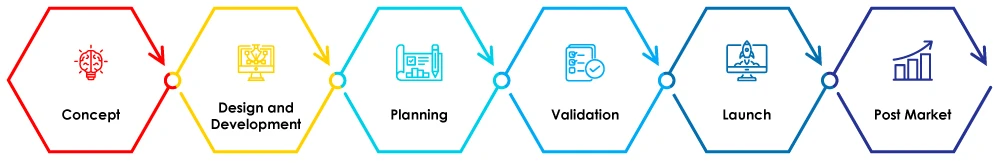
Abbildung 1 - Phasen des Lebenszyklus von Medizinprodukten
In diesem Blog werden wir die Bedeutung von QMS in der Design- und Entwicklungsphase des Lebenszyklus von Medizinprodukten diskutieren.
Design- und Entwicklungsphase im Lebenszyklus von Medizinprodukten
Die Entwurfs- und Entwicklungsphase ist eine der kritischsten Phasen im Lebenszyklus eines Medizinprodukts. In dieser Phase wird das Design des Geräts entwickelt und es werden Prototypen erstellt, gefolgt von Verifikations- und Validierungstests als Teil des Lebenszyklus eines Medizinprodukts.
Um sicherzustellen, dass das Medizinprodukt den regulatorischen Anforderungen, der Sicherheit, der Wirksamkeit und den Erwartungen der Anwender entspricht, ist die Implementierung eines Quality Management System (QMS) in der Entwurfs- und Entwicklungsphase des Lebenszyklus eines Medizinprodukts unerlässlich.
Die Dokumentation ist in der Entwurfs- und Entwicklungsphase von Medizinprodukten von entscheidender Bedeutung. Das QMS stellt sicher, dass die gesamte Dokumentation in Bezug auf Design und Entwicklung kontrolliert, verwaltet und dokumentiert wird.
Die Design History File (DHF) eine wichtige Datei/Aufzeichnung, die alle Unterlagen zum Design und zur Entwicklung des Geräts enthält. Die DHF liefert den Nachweis, dass das Gerätedesign den gesetzlichen Anforderungen entspricht.
Das DHF sollte die Dokumentation zu Entwurfseingaben, Entwurfsergebnissen, Entwurfsprüfungen, Entwurfsverifizierung, Validierung, Änderungen am Entwurf und Risikomanagement enthalten. Erfahren Sie hier mehr über das DHF.
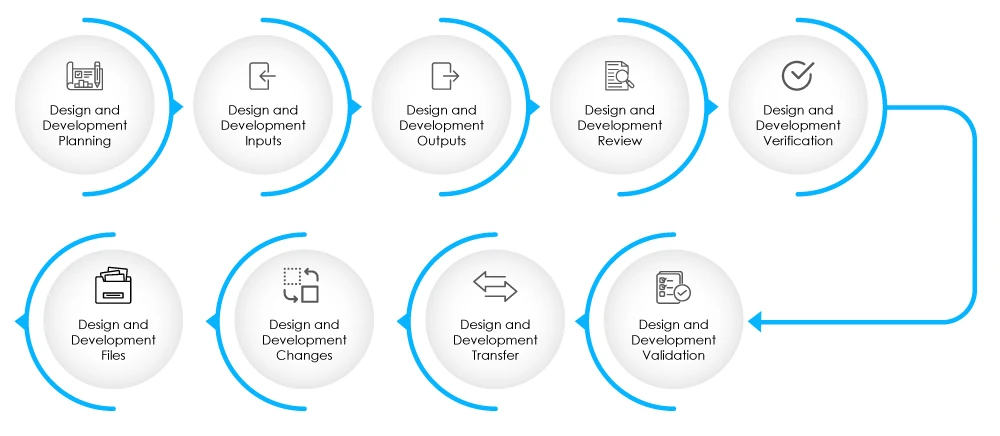
Abb. 2 - Phasen der Entwurfs- und Entwicklungsphase
Bewährte Praktiken für die Entwurfs- und Entwicklungsphase
- Erstellen Sie einen strukturierten Ansatz: Entwickeln Sie einen strukturierten Ansatz für die DHF-Entwicklung und -Verwaltung, der auf die spezifischen Bedürfnisse Ihrer Organisation zugeschnitten ist. Dieser Ansatz sollte klare Richtlinien, Verfahren und Arbeitsabläufe für die DHF-Entwicklung und -Verwaltung umfassen.
- Definieren und dokumentieren Sie die Design-Inputs: Definieren und dokumentieren Sie die Design-Inputs, einschließlich der Anforderungen und Spezifikationen für das Gerät, klar und deutlich. Dies kann dazu beitragen, dass die DHF umfassend und vollständig ist.
- Verwaltung von Entwurfsänderungen: Implementieren Sie einen robusten Änderungsmanagementprozess, der Verfahren zur Dokumentation, Bewertung und Genehmigung von Designänderungen umfasst. So kann sichergestellt werden, dass Änderungen ordnungsgemäß dokumentiert und hinsichtlich ihrer Auswirkungen auf die Sicherheit und Wirksamkeit des Produkts bewertet werden.
- Sicherstellung der Rückverfolgbarkeit: Entwickeln Sie eine Rückverfolgbarkeitsmatrix, die die Design-Inputs mit den Design-Outputs verbindet, und stellen Sie sicher, dass alle Design- und Entwicklungsaktivitäten ordnungsgemäß dokumentiert und aufgezeichnet werden. Dies kann dazu beitragen, dass die DHF rückverfolgbar ist und der Entscheidungsprozess gut dokumentiert wird.
- Gleichgewicht zwischen Innovation und Compliance: Entwickeln Sie eine Innovationskultur und stellen Sie gleichzeitig sicher, dass die Compliance-Anforderungen im Zusammenhang mit DHF, z. B. Designkontrollen und Risikomanagement, erfüllt werden. Dies kann durch die Entwicklung von Verfahren und Arbeitsabläufen erreicht werden, die Innovationen erleichtern und gleichzeitig sicherstellen, dass die gesetzlichen Anforderungen erfüllt werden.
- Dokumentenkontrolle einführen: Implementieren Sie Dokumentenkontrollverfahren, die sicherstellen, dass die DHF-Dokumente ordnungsgemäß kontrolliert werden, versionskontrolliert und für autorisiertes Personal zugänglich sind. Dies kann dazu beitragen, dass die DHF-Dokumente sicher sind und dass Änderungen ordnungsgemäß dokumentiert und genehmigt werden.
- Schulung des Teams: Stellen Sie sicher, dass das für die DHF-Entwicklung und -Verwaltung verantwortliche Team in Bezug auf die DHF-Anforderungen angemessen geschult ist und über das technische Fachwissen zur Entwicklung des Produkts verfügt. Dies kann durch regelmäßige Schulungen, Mentoring und die Einstellung von erfahrenen Fachleuten mit den erforderlichen Fähigkeiten und Kenntnissen erreicht werden.
Durch die Befolgung dieser bewährten Verfahren kann die Medizinprodukteindustrie die Einhaltung der gesetzlichen Vorschriften gewährleisten, die Sicherheit und Wirksamkeit ihrer Produkte fördern und ihren Wettbewerbsvorteil auf dem Markt wahren.
Zusammenfassend lässt sich sagen, dass die Einführung eines QMS bereits in der Entwurfs- und Entwicklungsphase entscheidend für den Erfolg in der stark regulierten Medizinprodukteindustrie ist. Durch die systematische Führung von Aufzeichnungen und die Einhaltung der behördlichen Anforderungen kann die Medizinprodukteindustrie sicherstellen, dass sie qualitativ hochwertige Produkte liefert und die Kundenzufriedenheit aufrechterhält.
Bei Freyr bieten wir QMS-Dienstleistungen an, um die Medizinprodukteindustrie dabei zu unterstützen, die regulatorischen Anforderungen in allen Phasen des Lebenszyklus von Medizinprodukten zu erfüllen.Kontaktieren Sieunsere QMS- und Regulierungsexperten, um mehr zu erfahren.









